Introduction
Nonalcoholic fatty liver disease (NAFLD) occurs in
individuals that have not consumed alcohol in amounts considered
harmful to the liver. The disease is characterized by
macrovesicular hepatic steatosis. There are two histological
patterns of NAFLD: Fatty liver alone and steatohepatitis.
Nonalcoholic steatohepatitis (NASH) is defined as lipid
accumulation in the liver with evidence of cellular damage,
inflammation and varying degrees of scarring or fibrosis (1).
The mechanisms underlying the increase in hepatic
fat accumulation are unclear and little information is available on
the time course of the development of hepatic steatosis. The
association between macrovesicular steatosis of the liver and
inflammatory changes and fibrosis in obese individuals has been
reported for several decades (2).
The spectrum of this condition ranges between silent nonalcoholic
fatty liver disease and chronic liver disease with complications.
Animal models indicate that the liver may accumulate lipids within
a few weeks or even a few days (3).
NAFLD is present in the majority of patients with
metabolic risk factors, including obesity and diabetes. Insulin
resistance (IR) is a key pathogenic factor of hepatic fat
accumulation. NAFLD exacerbates hepatic IR and precedes glucose
intolerance. Once hepatic steatosis occurs, additional factors,
including oxidative stress, mitochondrial dysfunction and
adipocytokines, may enhance hepatocellular damage (4).
NASH may progress to cirrhosis, particularly when
features of bridging fibrosis are present. No definite therapy is
currently available to reverse this pathological process (4). A number of studies have investigated
the efficacy of medications for the treatment of NASH; the drugs
studied include orlistat, metformin, vitamin E, lipid-lowering
drugs, pentoxifylline and pioglitazone (4,5). In
addition, thrombin inhibitors have been considered as a treatment
for liver fibrosis (6), and have
been found to decrease the lung collagen involved in pulmonary
fibrosis (7–8).
The current study aimed to examine the effects of
heparin, a thrombin inhibitor, on hepatic injury in an experimental
model of NASH and IR, via analyzing the effects on hepatic
inducible nitric oxide synthase (iNOS) expression and superoxide
generation.
Materials and methods
Drugs
Heparin sodium (Nile Pharma, Cairo, Egypt) was
administered subcutaneously to the mice in the herparin groups at a
dose of 10 μg/day (8).
Animals
A total of 80 male BALB/c mice (weight, 20–23 g;
age, 5–7 weeks) were obtained from the Animal Center of the
Research Unit at the Faculty of Medicine of Mansoura University
(Mansoura, Egypt). The experiment was performed in accordance with
the Guide for the Care and Use of Laboratory Animals, and the
experimental procedures were approved by the local Animal Care and
Use Committee of Mansoura University (Mansoura, Egypt). The animals
were housed in plastic cages with a 12-h light/dark cycle, with
free access to food and water throughout the experimental
period.
Experimental design
Following acclimatization for one week through
consumption of a basal diet, the mice were divided into four groups
(n=20) and fed the following diets for eight weeks. Control group
mice were fed a normal basal diet. The mice in the high fat (HF)
diet group were fed a diet containing 71% energy from fat, 11%
energy from carbohydrates and 18% energy from proteins (9). Mice in the HF diet + heparin-treated
group were administered 10 μg heparin sodium via daily subcutaneous
injections, following the initial consumption of the HF diet for
one week (second week overall) and for the remaining weeks
(6–8). Finally, mice in the heparin control
group were fed the normal basal diet, but administered heparin as
previously described.
Specimen collection and staining
Eight weeks from the start of the experiment, mice
were anesthetized with an intraperitoneal injection of sodium
pentobarbital (50 mg/kg body weight). Blood samples were collected
for biochemical analysis and the livers were harvested for iNOS
gene expression, histological and immunohistochemical
examinations.
Biochemical analysis
Serum levels of aspartate aminotransferase (AST) and
alanine aminotransferase (ALT) were measured using an automated
technique (Clinical Chemistry Analyzer), while serum glucose levels
were measured using a OneTouch® Ultra® Blood
Glucose meter (LifeScan, Inc., Milpitas, CA, USA). In addition,
serum insulin levels were measured with an enzyme-linked
immunosorbent assay (ELISA) kit (Crystal Chem, Inc., Downers Grove,
Chicago, IL, USA), according to the manufacturer’s instructions.
The level of hepatic triglyceride (TG) was measured using an
enzymatic method (Sigma-Aldrich, St. Louis, MO, USA), according to
the manufacturers’ instructions (10), while the hepatic hydroxyproline
content was estimated, as described in a study by Bergman and
Loxley (11). Hepatic superoxide
anion generation was also estimated using a previously described
method (12). The gene expression
levels of iNOS were determined in the livers of the aforementioned
groups by semi-quantitative polymerase chain reaction (PCR). In
addition, PCR was performed with the livers of a fifth group of
mice (n=12) that received high fat diet and subcutaneous injections
of a standard nonselective iNOS inhibitor, NG-monomethyl-L-arginine
(L-NMMA; Sigma-Aldrich), at a dose of 25 mg/kg/day (as with the
administration of the heparin regimen) (13) to inhibit the iNOS synthase
enzyme.
Total RNA was extracted from tissue specimens using
a column technique, according to the manufacturer’s instructions
(Vivantis Technologies, Subang Jaya, Malaysia). In total, 1 μg
total RNA was reverse transcribed to cDNA using a Maxima First
Strand cDNA Synthesis kit (Thermo Fisher Scientific, Ontario,
Canada). A total of 2 μl cDNA was amplified in a thermal cycler
instrument (Arktik Thermal Cycler; Thermo Fisher Scientific,
Pittsburgh, PA, USA) with 25 pmol each primer pair, 10 mM of
Tris-HCl, pH 8.3, 50 mM of KCl, 1.5 mM of MgCl2, 0.3 mM
of dNTP, 12.5 μl 2X Taq PCR Master mix (Qiagen, Inc.,
Valencia, CA, USA) and nuclease-free water in a total volume of 25
μl.
The primers designed for iNOS were 5′-CTCGGA
ACTGTAGCACAGCA3′ (sense) and 3′-GCACATCAAAGC GGCCATAG5′
(antisense). β-actin was included as a reference gene, and had
primer sequences of 5′-CAGGATTCCATA CCCAAGAAG-3′ (sense) and
3′-AACCCTAAGGCCAAC CGTG-5′ (antisense). The cycling parameters of
the PCR amplification were as follows: Initial denaturation at 95°C
for 5 min, followed by 35 cycles of denaturation at 94°C for 30
sec, annealing at 60°C for 30 sec and extension at 72°C for 45 sec,
which was followed by a final extension at 72°C for 5 min. PCR
products were electrophoresed in 2% agarose gel (Vivantis
Technologies) and visualized with an ethidium bromide stain
(Sigma-Aldrich). A 100-bp DNA ladder (Vivantis Technologies) was
applied to the first well to identify the molecular weight of the
samples. Gel images were captured and analyzed with a Gel
Documentation system (Bio-Rad, Hercules, CA, USA).
Histological procedure
Small pieces of liver were preserved in 10% formal
saline for ≥48 h, dehydrated in ascending grades of alcohol,
cleared in xylene, embedded in paraffin and sectioned into 5-μm
thick sections. The sections were stained with hematoxylin and
eosin to detect the histopathological changes, while Masson’s
trichrome and Sirius red stains were used to detect collagen. An
α-smooth muscle actin (α-SMA; Sigma-Aldrich, St.Louis, MO, USA)
immunohistochemical stain was used to detect activated Ito
cells.
Immunohistochemical procedure
A three-step indirect immunohistochemical technique
was performed on 5-μm formalin-fixed and paraffin-embedded
sections. Antigen retrieval was achieved by heating the sections in
a microwave oven at 560 W for 21 min in citrate buffer (pH 6.0).
The sections were subsequently treated with methanol containing
0.3% hydrogen peroxide for 15 min at room temperature in order to
inactivate the endogenous peroxidase. Nonspecific binding of the
secondary antibodies was minimized by incubation with 50% normal
goat serum in phosphate-buffered saline (PBS) for 20 min. Sections
were incubated with appropriate primary antibodies (α-SMA 1A4; 1/50
dilution) and diluted in PBS for 1 h in a humid chamber at room
temperature. All rinsing procedures and serum dilutions were
performed in PBS (pH 7.2–7.4). The detection kit used was a Dako
Cytomation LSAB™2 System-HRP for rabbit and mouse
(K0675; Dako Cytomation, Glostrup, Denmark). Positive reactions
were visualized by applying DAB+ liquid (K3468; Dako, Carpinteria,
CA, USA) for 5–10 min. Counterstaining with hematoxylin was
conducted for 2 sec and Aqueous Glycergel® Mounting
medium (C563; Dako) was used to mount the stained sections.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Analysis of variance followed by Tukey’s post-hoc test was used to
analyze the data, where P≤0.05 was considered to indicate a
statistically significant difference. All analyses were conducted
using SPSS 11.0 software for Windows (SPSS, Inc., Chicago, IL,
USA).
Results
Effects of heparin injection on serum
levels of ALT and AST
When compared with the control group, the serum
levels of ALT and AST were significantly higher in the HF diet
group (P≤0.05). The increment of these parameters was significantly
attenuated in the HF + heparin group (P≤0.05; Table I) when compared with the HF diet
group.
 | Table IEffect of heparin on various
parameters in a NASH mouse model. |
Table I
Effect of heparin on various
parameters in a NASH mouse model.
| Parameter | Control | HF diet | HF + heparin | Heparin
control |
|---|
| Serum insulin
(μU/ml) | 10.83±1.37 | 33.33±3.97a | 12.33±1.73b | 11.67±2.40b |
| Serum glucose
(mg/dl) | 102.50±6.40 |
262.50±18.97a | 114.33±7.01a,b | 105.17±8.02b,c |
| Superoxide
generation (pmol/min/mg protein) | 24.83±6.63 | 54.17±5.74a | 23.67±5.37b | 22.17±8.46b |
| TG (mg/g) | 31.00±5.41 | 131.67±7.16a | 43.83±9.90a,b | 30.83±5.65b,c |
| Hydroxyproline
(μmol/mg protein) | 2.27±1.51 | 7.40±1.53a | 2.93±0.71b | 2.52±0.86b |
| Serum ALT
(U/l) | 28.00±4.73 | 91.67±5.21a | 36.50±6.40a,b | 27.33±4.46b,c |
| Serum AST
(U/l) | 74.00±6.35 |
154.50±12.63a | 78.00±9.81b | 73.67±9.96b |
| HOMA | 2.76±0.49 | 21.54±2.53a | 3.47±0.44b | 3.01±0.60b |
| Body weight
(g) | 19.50±5.30 | 23.30±7.10 | 21.50±6.30 | 20.60±5.90 |
Effects of heparin injection on oxidative
stress, hepatic TG and hepatic fibrosis
As shown in Table
I, the levels of superoxide free radicals, hepatic
hydroxyproline, TGs and serum insulin were significantly higher in
the HF diet group when compared with the control group (P≤0.05).
This increase was significantly reduced in the heparin-treated
groups (HF + heparin only; P≤0.05) when compared with the HF diet
group.
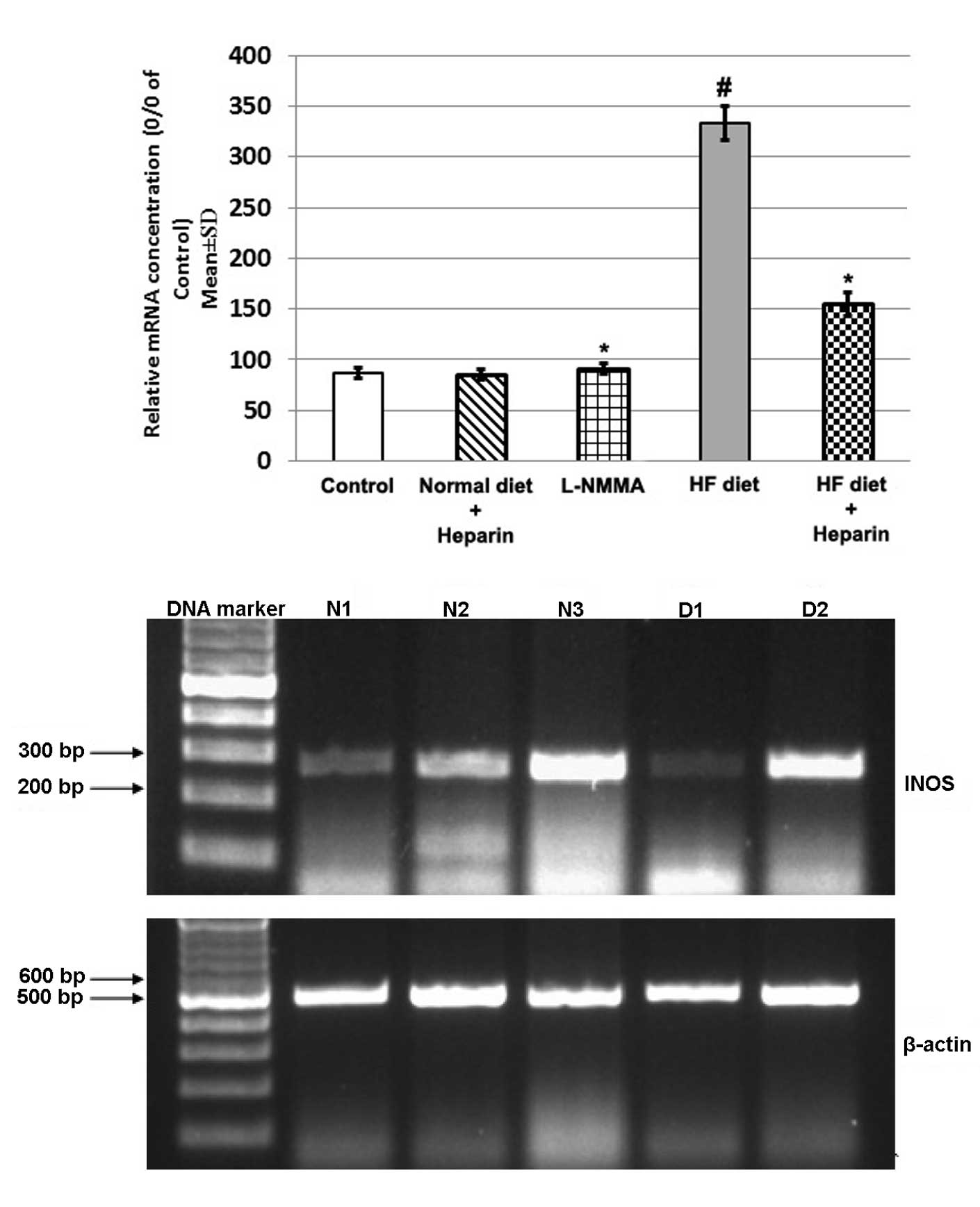
Effect of heparin on the mRNA expression
of iNOS
Hepatic expression of iNOS was higher in the HF diet
group compared with the control group. However, heparin treatment
decreased iNOS expression when compared with the L-NMMA-treated HF
diet group (Fig. 1).
Histopathological observations of
NAFLD
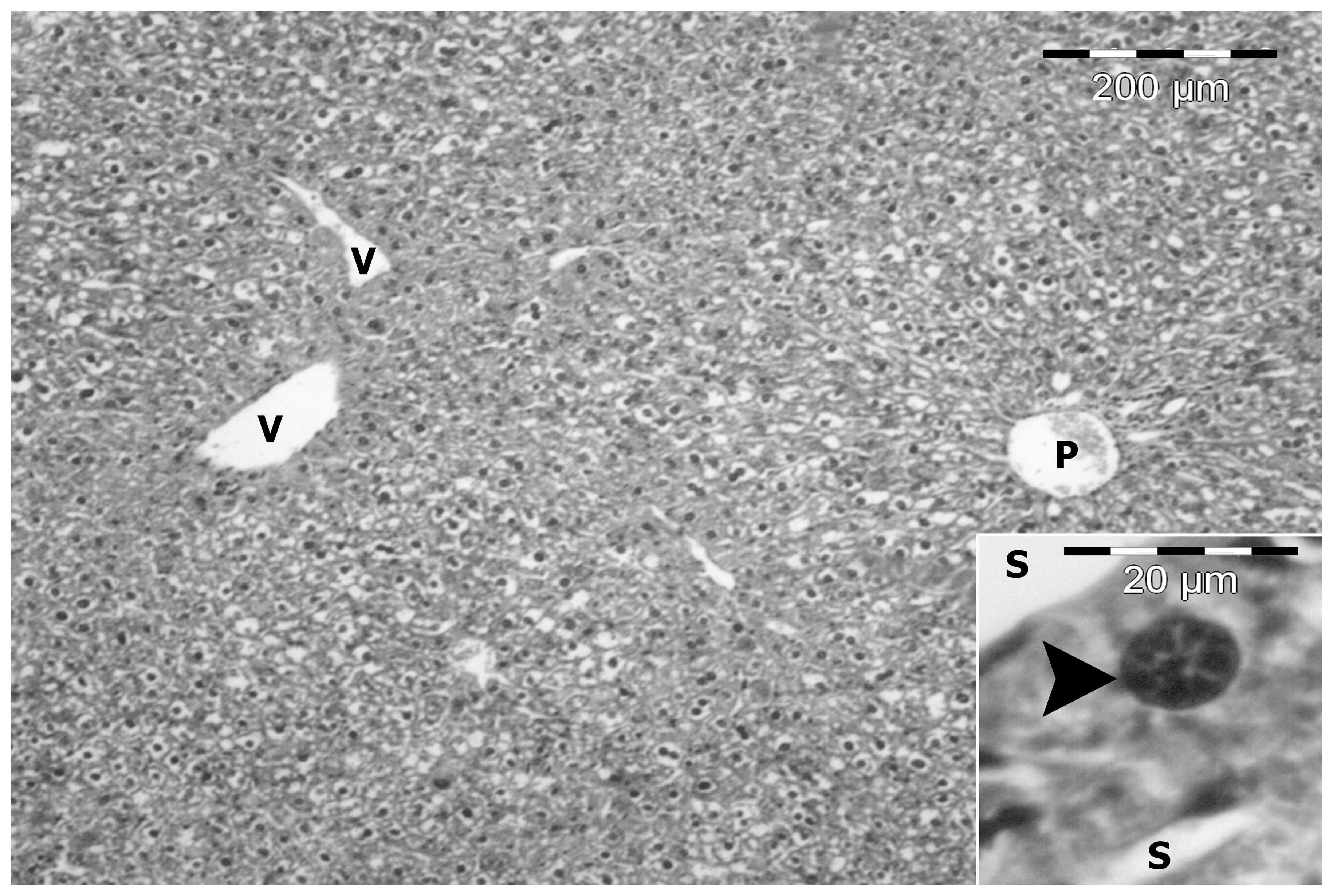
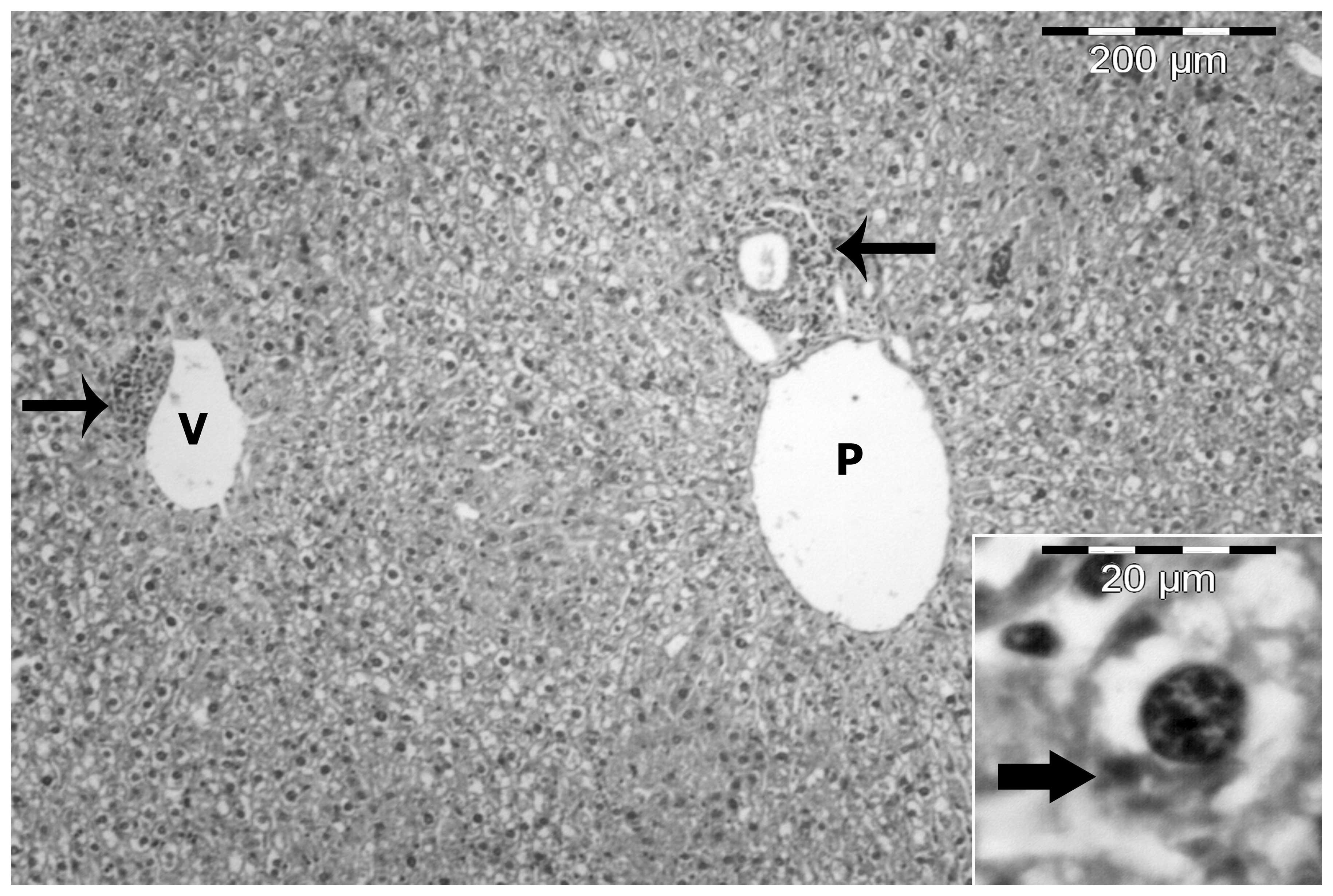
In the control group, livers were composed of a
number of lobules consisting of thin-walled central veins from
which cords of hepatocytes radiated towards the lobule periphery
and alternated with narrow irregular blood sinusoids. Around the
periphery of each lobule, branches of the hepatic artery, hepatic
portal vein and bile duct were present forming the portal tract.
The hepatocytes were polygonal with a large vesicular nucleus
(Fig. 2).
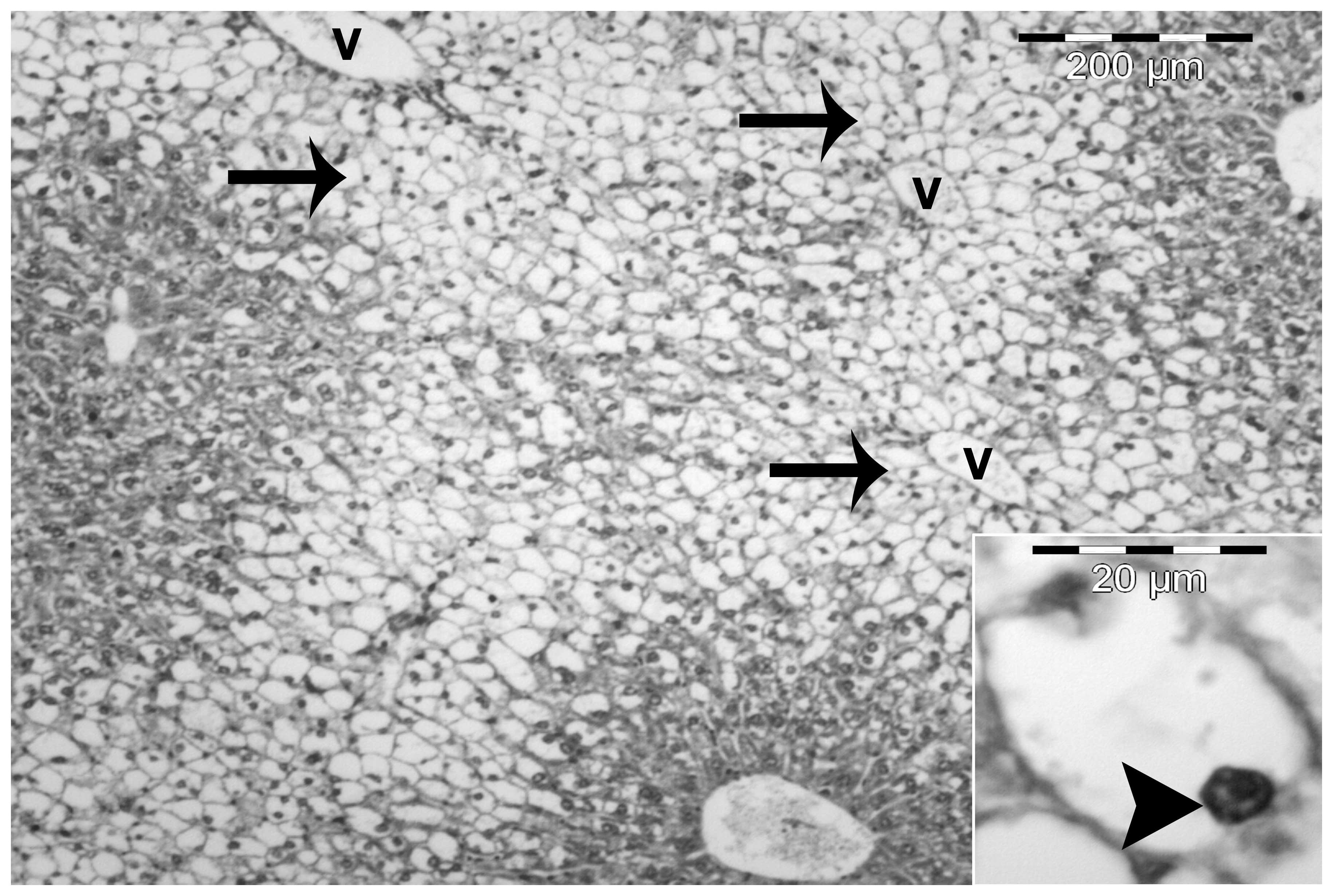
The most important histological characteristic of
the HF diet group was the presence of severe macrovesicular and
microvesicular steatosis in the hepatocytes. The steatosis was
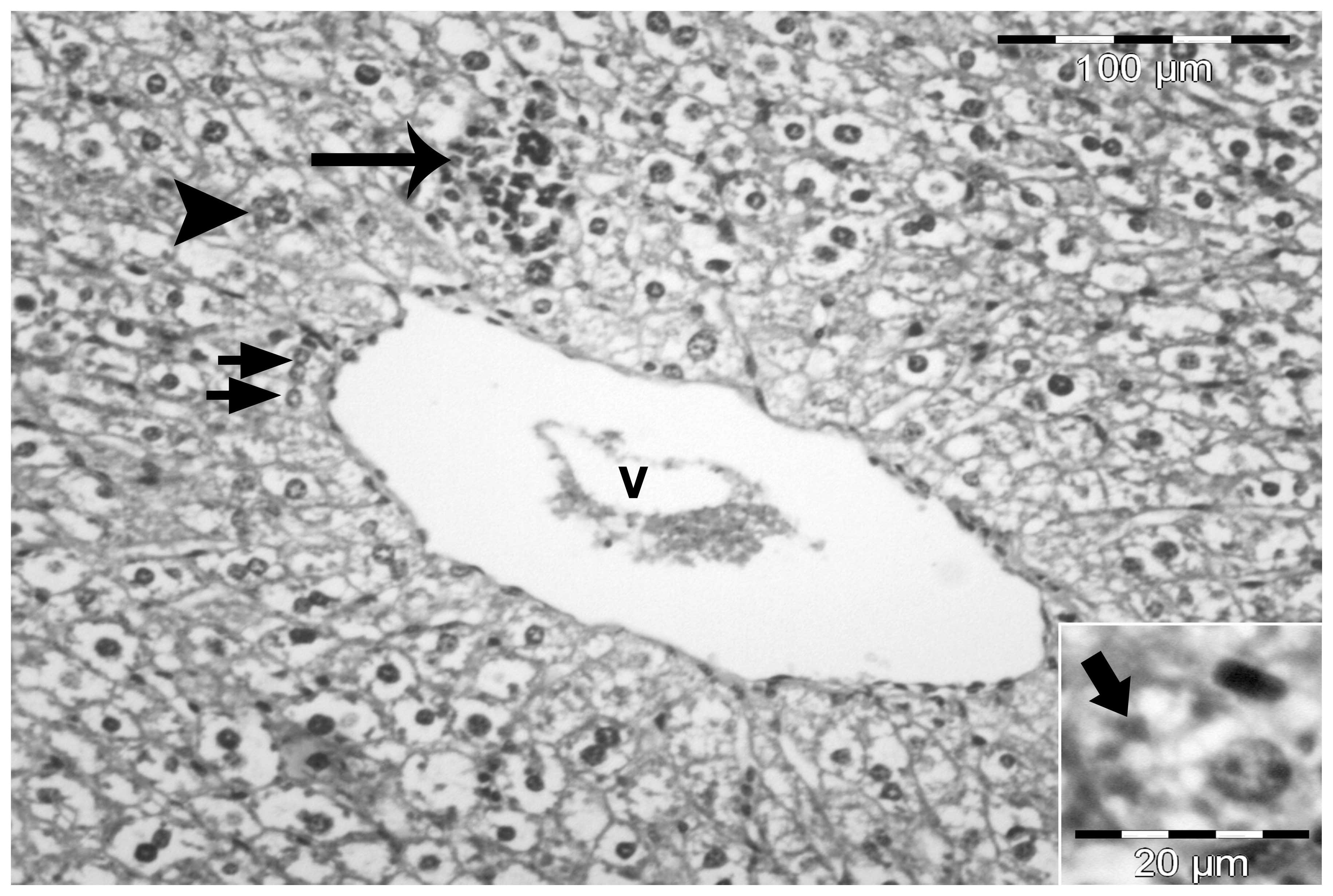
predominantly centrilobular and macrovesicular (Fig. 3). However, in certain sections,
steatosis was observed throughout the lobule. Macrovesicular
steatosis was characterized by large vacuoles occupying almost the
entire cytoplasm and forcing the nucleus to the periphery of the
cell. By contrast, microvesicular steatosis was characterized by
multiple small lipid vacuoles with the nucleus located in the
center of the cell (Figs. 3 and
4).
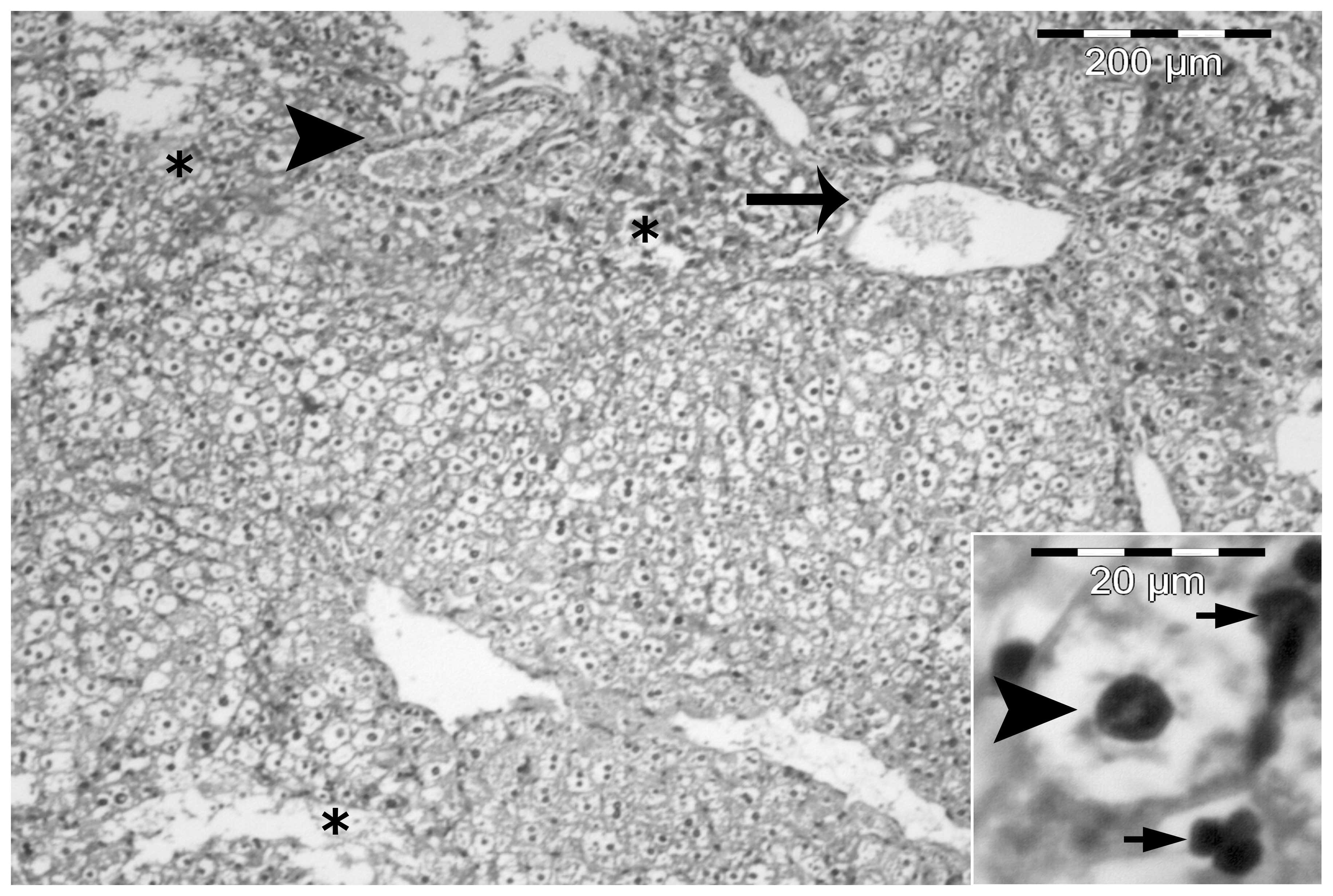
A number of animals developed NASH, where steatosis
was accompanied by mild intralobular and periportal inflammation
with prominent hepatocellular ballooning (Fig. 4). Certain sections also
demonstrated fat cysts and lipogranulomas, glycogenated nuclei and
cells containing Mallory’s hyaline (Fig. 5). In the heparin-treated group,
steatosis and necroinflammatory activity was minimal (Fig. 6).
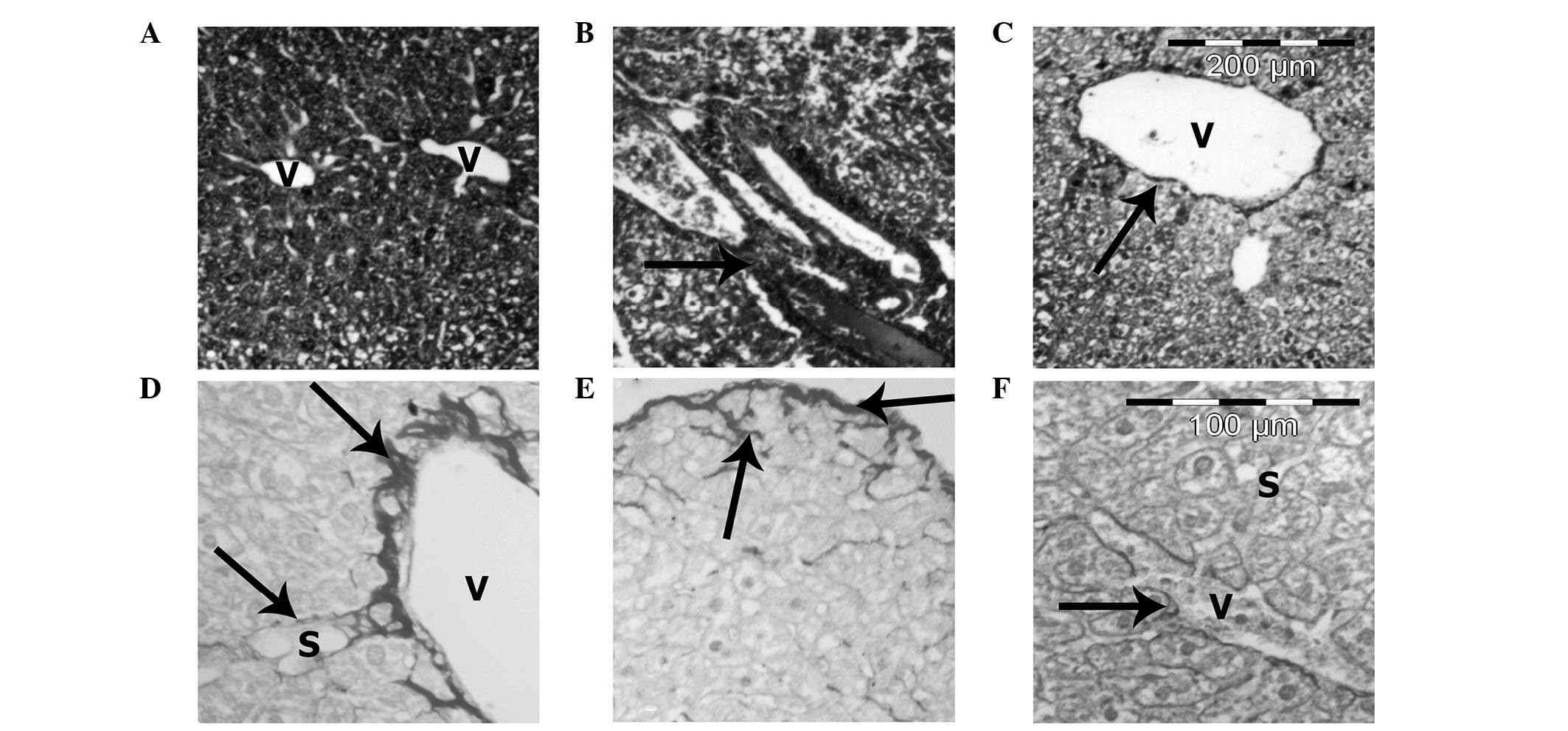
Heparin treatment improved liver
fibrosis
Masson’s trichrome and Sirius red stains for
collagen revealed minimal fibrosis in the control group (Fig. 7A). However, the extent of fibrosis
increased in the HF diet group, particularly in the perisinusoidal
area in zone 3 and in the periportal region. A number of animals
also demonstrated bridging fibrosis and pericellular fibrosis
(Fig. 7B, D and E). However,
heparin treatment decreased the extent of fibrosis when compared
with the HF diet group (Fig. 7C and
F).
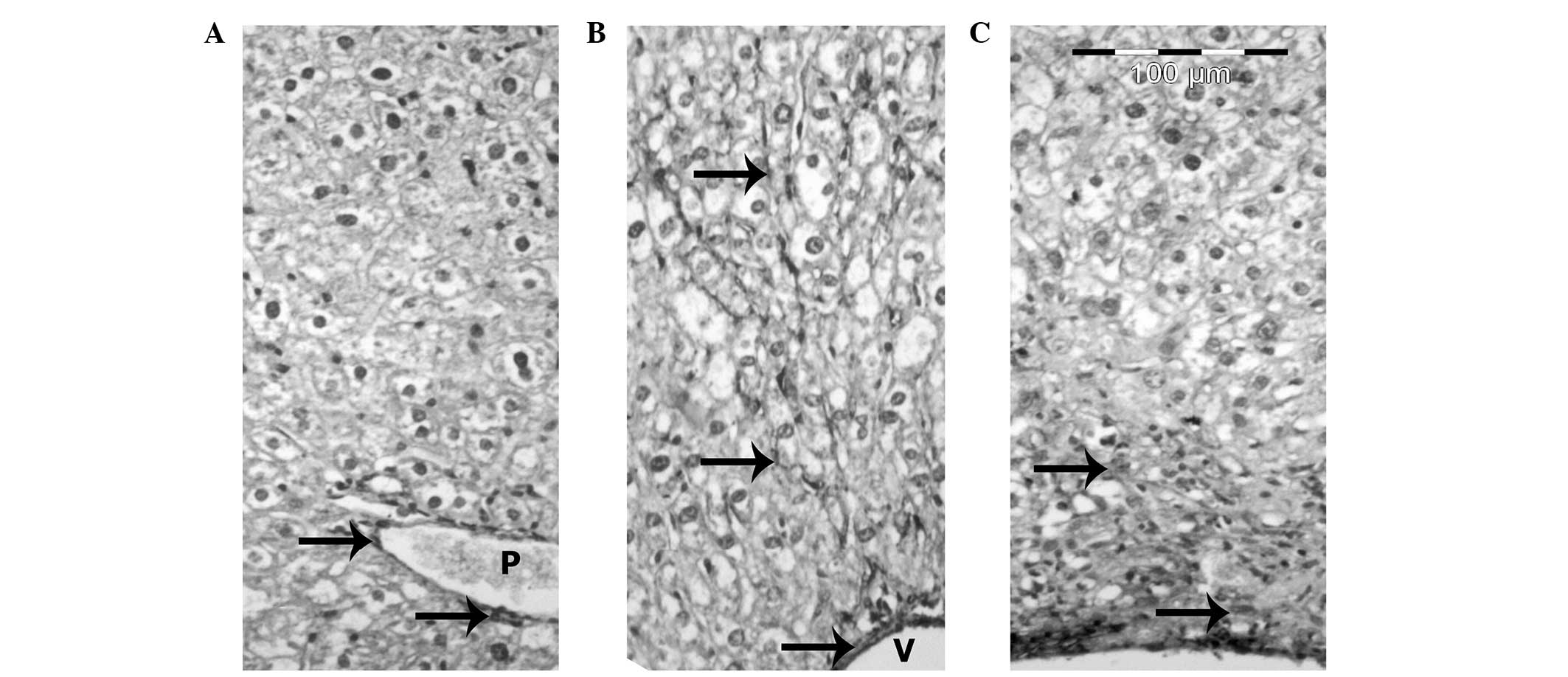
Immunohistochemistry
In the liver sections of the control mice, a
positive reaction between the smooth muscle cells of the terminal
and sublobular venous blood vessels was observed (Fig. 8A). In the HF diet group, Ito cells
reacted positively with the α-SMA antibody in the areas of fibrosis
and in the walls of the central veins, as well as in the septal
connective tissue (Fig. 8B). The
livers of mice from the HF diet + heparin-treated group revealed
fewer α-SMA positive cells compared with the HF diet group
(Fig. 8C).
Discussion
NAFLD is present in the majority of patients with
metabolic and/or multiple risk factors, including obesity, diabetes
and hypertension. There are two histological patterns of NAFLD:
Fatty liver and steatohepatitis. NASH is defined as lipid
accumulation associated with cell damage, inflammatory changes and
fibrosis (1). NASH is a serious
problem, with almost 25% of cases progressing to cirrhosis and
subsequent complications of portal hypertension, liver failure and
hepatocellular carcinoma (14).
In the present study, HF diet-fed mice were used to
examine the effect of heparin on iNOS enzyme expression. The HF
diet induced a pathology similar to human NASH, including
steatosis, hepatic inflammation and fibrous tissue formation. Mice
fed a HF diet for eight weeks also developed hyperglycemia and
hyperinsulinemia, which mimicked the human metabolic syndrome
process. These experimental protocols were selected in accordance
with the study by Lieber et al (9), who reported that rats fed a HF diet
ad libitum for three weeks demonstrated IR, as well as
marked panlobular steatosis, increased hepatic lipid concentrations
and oxidative damage.
The pathogenesis of steatohepatitis caused by
metabolic factors is not fully understood; however, the induction
of fat into the liver is essential. In experimental steatohepatitis
caused by a lipogenic diet, hepatic steatosis represents the
hepatic part of the metabolic syndrome disorder and is accompanied
by IR and elevated TGs (2,15).
The mechanism responsible for the increase in
hepatic lipid accumulation is uncertain and little information is
available regarding the time course of the development of hepatic
steatosis. It has been hypothesized that fatty liver may result
from increased delivery of free fatty acids (FFAs), impaired
hepatic fatty acid oxidation and/or impaired synthesis or secretion
of very low-density lipoproteins (16). Among these factors, the increased
flow of FFAs to the liver is considered the most important
(17). An increased rate of
lipolysis and FFA abundance are the causes of oxidative stress in
the liver. FFA oxidation generates oxidative stress and subsequent
lipid peroxidation, which may be the mediator of subsequent
inflammatory processes (18).
Oxidative stress is a mechanism of hepatocellular
injury in steatohepatitis and is associated with the accumulation
of lipid peroxidation products, elevation of proinflammatory
cytokines and mitochondrial dysfunction (18–19).
However, excess hepatic fat may lead to hepatic IR via the release
of cytokines from Kupffer cells, leading to hepatocellular
inflammation, reduced endothelial nitric oxide (eNO) signaling and
the development of IR, which is a risk factor for liver fibrosis in
steatohepatitis (20).
Excessive circulating FFAs exert deleterious effects
on mitochondria and insulin signaling via protein kinase C
activation and the inhibition of insulin receptor tyrosine kinase
activity, leading to systemic IR (21).
The hepatic fibrosis observations in the present
study, which resulted from TG accumulation and steatohepatitis,
were consistent with those reported in the study by Poniachik et
al (22), who concluded that
the degree of steatosis in the liver biopsy is a risk factor for
the development of fibrosis. In addition, Larter et al
(23) revealed that in a
nutritional model of steatohepatitis, accumulation of TGs occurs
despite substantial suppression of lipogenesis and the induction of
TG synthesis genes; these observations support the lipotoxicity
mechanism involved in the liver injury component of metabolic
syndrome.
Furthermore, the origin of the reactive oxygen
species increase in NASH has been investigated. Reduced superoxide
scavenging and glutathione replenishment may be involved in
steatohepatitis, and antioxidant therapy may have a beneficial
therapeutic effect for this purpose (24–25).
Administration of a HF diet in mice induces the
proinflammatory activation of Kupffer cells, which is associated
with reduced liver endothelial NOS activity. Kupffer cells produce
the free radical, NO, from iNOS when they are stimulated by
inflammatory cytokines or lipopolysaccharide (13–26).
Liu and Huang (27)
supported the association between obesity-induced IR and the
reduction in the eNO vascular content that causes a predisposition
to increased endothelial inflammation, thrombosis and
vasoconstriction. Furthermore, Tateya et al (28) revealed that a HF diet induces liver
inflammation and IR. The authors determined that a reduction in eNO
signaling leads to increased inflammation, which is inhibited by
increasing eNOS activity.
Immunohistochemical examination of the livers of the
mice in the HF diet group demonstrated increased expression levels
of α-SMA in the perisinusoidal stellate cells in areas of fibrosis
around the portal spaces and fibrous septa. These results have been
reported during the course of the development of hepatic fibrosis
in humans and rats (29–32). Activated hepatic stellate cells
synthesize tissue inhibitors of matrix metalloproteinase 1 and 2,
which inhibit interstitial collagenase activity and contribute to
the accumulation of extracellular matrix proteins; the latter of
which is involved in the progression of chronic hepatic fibrosis
(33–36).
NASH therapy remains under investigation and no
definite therapy is currently available to reverse the cirrhotic
pathological process (2).
Previously, heparin was used in the management of vascular
thrombosis. Heparin is a highly sulfated polysaccharide isolated
from mast cells, which has been described as having antioxidant and
antifibrotic activities. The drug has been used in the treatment of
hepatic cholestasis and fibrosis (6,7,37).
In the present study, subcutaneous injections of heparin into mice
fed a HF diet for eight weeks was shown to improve liver enzyme
function, IR and hepatic fibrosis via the prevention of hepatic
inflammation, improved IR and decreased signaling of iNOS. Heparin
is known to have biological effects that are not associated with
anticoagulant activity, including potential anti-inflammatory,
immunomodulatory and antitumor effects. Certain studies have
demonstrated that these actions may be hindered by the effect of
heparin on hemostasis; however, not in the dosage used in the
present study (6,8,37).
The clinical benefit of heparin on chronic
inflammatory disease has been previously studied. Heparin controls
inflammation through trapping and neutralizing the inflammatory
pathological proteins involved in the inflammatory process. This
ability may be the result of its large polysaccharide chemical
structure that binds to the different inflammatory mediators and
cytokines, including antithrombin, heparin binding proteins and
adhesion molecules (37). Mu et
al concluded that heparin ameliorates lung injury and causes
pulmonary vascular remodeling via the inhibition of iNOS expression
(38–39). Furthermore, Ding et al
(40) revealed that unfractionated
heparin reduces inflammation and cytokine expression in endotoxemic
mice. In addition, Saï et al (8) demonstrated that heparin attenuates
low-dose streptozotocin-induced immune diabetes in mice in a dose
similar to the dose used in the present study, but for a period of
almost three weeks, as well as inhibiting the β-cell binding of
T-splenocytes in vitro. The authors hypothesized that
heparin at this dosage exhibits a variety of functions beyond an
anticoagulant effect, without inducing the side effect of blood
loss.
Tokuyama et al (41) revealed that diabetic patients who
were administered insulin lispro and heparin treatment achieved
improved glycemic control without any adverse effects. In addition,
Shah and Shah demonstrated that heparin preserves hepatic function
and reduces the severity of hepatic fibrogenesis following six
weeks of treatment. Thus, heparin inhibits thrombin in vivo
and in vitro. Thrombin is responsible for stimulating the
proliferation of hepatic stellate cells (6). It interacts with specific cellular
upregulated receptors that are located not only in the liver, but
also in the lungs, resulting in the accumulation of collagen and
fibrosis. Heparin is involved in the inhibition of the coagulation
system and prevents acute liver injury from a hepatotoxic dose of
lipopolysaccharide (6,42).
In conclusion, heparin administration improves the
liver histopathology of mice receiving a HF diet. This effect
occurs via the downregulation of the iNOS gene and the suppression
of hepatic TG content. The anti-inflammatory mechanism of heparin
should be determined in future studies.
Acknowledgements
The authors thank the Experimental Medical Research
Center (Faculty of Medicine, Mansoura University) for their
significant contribution to the experimental care and biochemical
analysis, and the Nile Center for Research (in particular Dr Thoria
Medhat and her supervisor Dr Mahmoud Zakaria) for assistance with
semi-quantitative PCR.
References
|
1
|
Brunt EM, Neuschwander-Tetri BA, Oliver D,
Wehmeier KR and Bacon BR: Nonalcoholic steatohepatitis: histologic
features and clinical correlations with 30 blinded biopsy
specimens. Hum Pathol. 35:1070–1082. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marchesini G, Brizi M, Morselli-Labate AM,
Bianchi G, Bugianesi E, McCullough AJ, Forlani G and Melchionda N:
Association of nonalcoholic fatty liver disease with insulin
resistance. Am J Med. 107:450–455. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Diehl AM: Lessons from animal models of
NASH. Hepatol Res. 33:138–144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kita Y, Takamura T, Misu H, Ota T, Kurita
S, Takeshita Y, Uno M, Matsuzawa-Nagata N, et al: Metformin
prevents and reverses inflammation in a non-diabetic mouse model of
nonalcoholic steatohepatitis. PLoS One. 7:e430562012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCullough AJ: Thiazolidinediones for
nonalcoholic steatohepatitis - promising but not ready for prime
time. N Engl J Med. 355:2361–2363. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shah B and Shah G: Antifibrotic effect of
heparin on liver fibrosis model in rats. World J Gastrointest
Pharmacol Ther. 3:86–92. 2012.PubMed/NCBI
|
|
7
|
Howell M, Mohun TJ and Hill CS: Xenopus
Smad3 is specifically expressed in the chordoneural hinge,
notochord and in the endocardium of the developing heart. Mech Dev.
104:147–150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saï P, Pogu S and Ouary M: Heparin
attenuates low-dose streptozotocin-induced immune diabetes in mice
and inhibits the beta-cell binding of T-splenocytes in vitro.
Diabetologia. 34:212–217. 1991.PubMed/NCBI
|
|
9
|
Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q,
Ren C, Ponomarenko A and Decarli LM: Model of nonalcoholic
steatohepatitis. Am J Clin Nutr. 79:502–509. 2004.PubMed/NCBI
|
|
10
|
Bligh EG and Dyer WJ: A rapid method of
total lipid extraction and purification. Can J Biochem Physiol.
37:911–917. 1959. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bergman M and Loxley R: The determination
of hydroxyproline in urine hydrolysates. Clin Chim Acta.
27:347–349. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang HD, Pagano PJ, Du Y, Cayatte AJ,
Quinn MT, Brecher P and Cohen RA: Superoxide anion from the
adventitia of the rat thoracic aorta inactivates nitric oxide. Circ
Res. 82:810–818. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiemermann C, Ruetten H, Wu CC and Vane
JR: The multiple organ dysfunction syndrome caused by endotoxin in
the rat: attenuation of liver dysfunction by inhibitors of nitric
oxide synthase. Br J Pharmacol. 116:2845–2851. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ekstedt M, Franzén LE, Mathiesen UL,
Thorelius L, Holmqvist M, Bodemar G and Kechagias S: Long-term
follow-up of patients with NAFLD and elevated liver enzymes.
Hepatology. 44:865–873. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chitturi S and Farrell GC:
Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis.
21:27–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jansen PL: Non-alcoholic steatohepatitis.
Eur J Gastroenterol Hepatol. 16:1079–1085. 2004. View Article : Google Scholar
|
|
17
|
McClain CJ, Mokshagundam SP, Barve SS, et
al: Mechanisms of non-alcoholic steatohepatitis. Alcohol. 34:67–79.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonnefont-Rousselot D, Ratziu V, Giral P,
Charlotte F, Beucler I and Poynard T; Lido Study Group. Blood
oxidative stress markers are unreliable markers of hepatic
steatosis. Aliment Pharmacol Ther. 23:91–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Albano E: Free radical mechanisms in
immune reactions associated with alcoholic liver disease. Free
Radic Biol Med. 32:110–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim F, Pham M, Maloney E, Rizzo NO, Morton
GJ, Wisse BE, Kirk EA, Chait A and Schwartz MW: Vascular
inflammation, insulin resistance, and reduced nitric oxide
production precede the onset of peripheral insulin resistance.
Arterioscler Thromb Vasc Biol. 28:1982–1988. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guilherme A, Virbasius JV, Puri V and
Czech MP: Adipocyte dysfunctions linking obesity to insulin
resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 9:367–377.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Poniachik J, Mancilla C, Contreras J,
Csendes A, Smok G, Cavada G, Rojas J, et al: Obesity: risk factor
for steatohepatitis and hepatic fibrosis. Rev Med Chil.
130:731–736. 2002.(In Spanish).
|
|
23
|
Larter CZ, Yeh MM, Haigh WG, Williams J,
Brown S, Bell-Anderson KS, Lee SP and Farrell GC: Hepatic free
fatty acids accumulate in experimental steatohepatitis: role of
adaptive pathways. J Hepatol. 48:638–647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Laurent A, Nicco C, Tran Van Nhieu J,
Borderie D, Chéreau C, Conti F, et al: Pivotal role of superoxide
anion and beneficial effect of antioxidant molecules in murine
steatohepatitis. Hepatology. 39:1277–1285. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
von Montfort C, Matias N, Fernandez A,
Fucho R, Conde de la Rosa L, Martinez-Chantar ML, et al:
Mitochondrial GSH determines the toxic or therapeutic potential of
superoxide scavenging in steatohepatitis. J Hepatol. 57:852–859.
2012.PubMed/NCBI
|
|
26
|
Böhm T, Berger H, Nejabat M, Riegler T,
Kellner F, Kuttke M, Sagmeister S, Bazanella M, et al: Food-derived
peroxidized fatty acids may trigger hepatic inflammation: a novel
hypothesis to explain steatohepatitis. J Hepatol. 59:563–570.
2013.PubMed/NCBI
|
|
27
|
Liu VW and Huang PL: Cardiovascular roles
of nitric oxide: a review of insights from nitric oxide synthase
gene disrupted mice. Cardiovasc Res. 77:19–29. 2008.PubMed/NCBI
|
|
28
|
Tateya S, Rizzo NO, Handa P, et al:
Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell
activation and hepatic insulin resistance induced by high-fat
feeding. Diabetes. 60:2792–2801. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Friedmann SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boisclair J, Doré M, Beauchamp G,
Chouinard L and Girard C: Characterization of the inflammatory
infiltrate in canine chronic hepatitis. Vet Pathol. 38:628–635.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bataller R and Brenner D: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar
|
|
32
|
Mekonnen GA, Ijzer J and Nederbragt H:
Tenascin-C in chronic canine hepatitis: immunohistochemical
localization and correlation with necro-inflammatory activity,
fibrotic stage, and expression of alpha-smooth muscle actin,
cytokeratin 7, and CD3+ cells. Vet Pathol. 44:803–813.
2007. View Article : Google Scholar
|
|
33
|
Bedossa P and Paradis V: Liver
extracellular matrix in health and disease. J Pathol. 200:504–515.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guyot C, Lepreux S, Combe C, Doudnikoff E,
Bioulac-Sage P, Balabaud C and Desmoulière A: Hepatic fibrosis and
cirrhosis: the (myo)fibroblastic cell subpopulations involved. Int
J Biochem Cell Biol. 38:135–151. 2006.PubMed/NCBI
|
|
35
|
Hinz B, Phan SH, Thannickal VJ, Galli A,
Bochaton-Piallat M and Gabbiani G: The myofibroblast: one function,
multiple origins. Am J Pathol. 170:1807–1816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ijzer J, Roskams T, Molenbeek RF, Ultee T,
Penning LC, Rothuizen J and van den Ingh TS: Morphological
characterization of portal myofibroblasts and hepatic stellate
cells in the normal dog liver. Comp Hepatol. 5:72006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lever R and Page CP: Non-anticoagulant
effects of heparin: an overview. Handb Exp Pharmacol. 207:281–305.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mu E, Ding R, An X, Li X, Chen S and Ma X:
Heparin attenuates lipopolysaccharide-induced acute lung injury by
inhibiting nitric oxide synthase and TGF-β/Smad signaling pathway.
Thromb Res. 129:479–485. 2012.
|
|
39
|
Horstman DJ, Fischer LG, Kouretas PC,
Hannan RL and Rich GF: Role of nitric oxide in heparin-induced
attenuation of hypoxic pulmonary vascular remodeling. J Appl
Physiol (1985). 92:2012–2018. 2002.PubMed/NCBI
|
|
40
|
Ding R, Zhao D, Guo B, Zhang Z and Ma X:
Treatment with unfractionated heparin attenuates coagulation and
inflammation in endotoxemic mice. Thromb Res. 128:e160–e165. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tokuyama Y, Nozaki O and Kanatsuka A: A
patient with subcutaneous-insulin resistance treated by insulin
lispro plus heparin. Diabetes Res Clin Pract. 54:209–212. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fiorucci S, Antonelli E, Distrutti E,
Severino B, Fiorentina R, Baldoni M, et al: PAR1 antagonism
protects against experimental liver fibrosis. Role of proteinase
receptors in stellate cell activation. Hepatology. 39:365–375.
2004. View Article : Google Scholar : PubMed/NCBI
|