Introduction
Paroxysmal kinesigenic dyskinesia (PKD), also known
as paroxysmal exercise-induced dyskinesia syndrome, is an autosomal
dominant genetic disease, the incidence rate of which is ~1/150,000
(1). The clinical features are
sudden exercise-induced dance-like movements of the limbs or torso,
throwing disorders, stiffness or numbness, dystonia and other
actions (2). PKD is mostly onset
in childhood and increases in prepubescence; however, the symptoms
are spontaneously relieved in adulthood and disappear at an age of
30–40 years (3). The patients have
clear consciousness at onset, and exhibit normal
electroencephalograms during seizures and the interictal period,
which differentiates it from epilepsy. Patients with PKD are very
sensitive to antiepileptic drugs; small doses of carbamazepine or
phenytoin can effectively control the seizures of PKD, and the
symptoms may disappear completely in certain patients after
treatment with drugs for three days (4).
Since it was first reported in 1967 (5), it has been confirmed that there are
two pathogenic gene regions in PKD, specifically 16p11.2-q12.1 and
16q13-q22.1 (6,7). There may be a third pathogenic region
in PKD; however, this is not on chromosome 16 (8). Nevertheless, the causative gene has
not been cloned. In 2011, Chen et al collected eight
families with PKD and discovered using whole exon sequencing
combined with Sanger sequencing that all eight families were
carrying PRRT2 gene mutations, while no PRRT2 mutations were found
in the 1,000 cases of the normal control group (9). Therefore, Chen et al
identified PRRT2 as a causative gene of PKD, which was confirmed in
subsequent studies (10–12). However, there is little literature
concerning PKD, and genetically confirmed cases of PKD are rarely
reported. In the present study, nine cases of clinically diagnosed
PKD were collected from 2007 onwards, and the genes of these
patients were sequenced by the Sanger method. The incidence of
PRRT2 mutations in the patients was determined to investigate PRRT2
as a pathogenic gene in PKD.
Materials and methods
Subjects
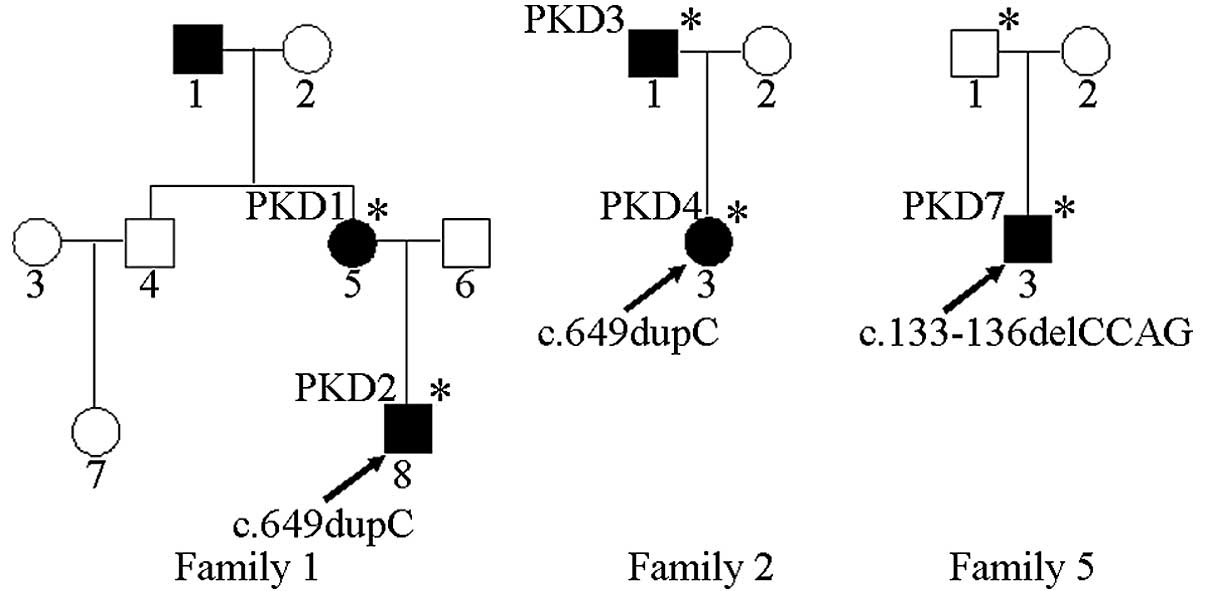
The study included nine patients with PKD (cases PKD
1–9) from seven different pedigrees (families 1–7). These included
six male cases (PKD 2, 3, 5 and 7–9) and three female cases (PKD 1,
4 and 6). Four patients had a family history (PKD 1–4) and the
other five were sporadic cases. Family 1 had eight members, three
of whom had a history of PKD, but the patient 1 refused a blood
sample and thus it was not possible to determine whether that
patient carried PRRT2 mutations. The family 2 proband exhibited
involuntary twisting of the body and his father mainly complained
of sudden movement. The remaining five probands and their parents
were asymptomatic and classified as sporadic cases. The present
study was approved by the Ethics Committee of Zhengzhou Children’s
Hospital (Zhengzhou, China). All patients provided informed
consent.
Blood sample collection
Venous blood samples (3 ml) were obtained for
anticoagulation by sodium citrate. They were stored in a
refrigerator at 4°C for short periods, and frozen at −20°C for
long-term preservation.
DNA extraction
The QIAamp blood DNA extraction kit (Qiagen, Hilden,
Germany) was used to extract DNA from the blood samples. The DNA
was dissolved with 1X AE solution (Qiagen), and stored at −20°C for
the long-term.
Polymerase chain reaction (PCR)
Five PRRT2 primers were used according to the
literature (9), and are shown in
(Table I). Four exons including
exon-intron joints (≥20 bp) were amplified. The PCR annealing
temperature was 66°C (exon 1) or 60°C (exon 2a, 2b, 2c, 3–4).
 | Table IPRRT2 gene forward and reverse
primers. |
Table I
PRRT2 gene forward and reverse
primers.
| Exons | Forward primers
(5′→3′) | Reverse primers
(5′→3′) |
|---|
| Exons 1 |
TTGCCTGGGTAACGCGTGGCT |
ACACCCGCATTCCCGTGCAGT |
| Exons 2a | CAATT
GGGCCTGCAGTGCTGAG |
GGTTTGGACACTGTTTCTTGGCAT |
| Exons 2b |
GGAGGGGAATCAAAGGCCAACTG |
TCAACCAGCTGCTGCAGCACTC |
| Exons 2c |
GAAAAGCAAGAGAATGGGGCAGTG |
GATTACTCCAGAGGCTCTATTGCAG |
| Exons 3–4 |
TTCTGGATGACTTTTCCACCTGAT |
CAACAGGAAGAAAAGTCTTGGGAT |
Sequencing
To 5 μl PCR product was added 0.3 μl shrimp
alkaline/heat-sensitive alkaline phosphatase [Promega (Beijing)
Biotech Co., Ltd., Beijing, China], 0.2 μl exonuclease [New England
Biotechnology (Beijing) Co., Ltd., Beijing, China] and 2.5 μl
ddH2O for purification. The mixture was placed in a 37°C
water bath for 1 h, then in a 80°C water bath for 15 min. The 3 μl
purified PCR product that was obtained was added to 1 μl 3.2 pmol/l
unidirectional primer and 1 μl BigDye working fluid (Beijing Yue
Wei Gene Technology Co., Ltd., Beijing, China) for forward
sequencing reaction. The reaction conditions were: 96°C
denaturation for 1 min; 94°C denaturation for 10 sec, 50°C
annealing for 5 sec and 60°C extension for 4 min, for 25 cycles;
followed by 60°C extension for 10 min. Following the sequencing
reaction, 50 μl 75% ethanol mixture was added and the resultant
mixture was stored at −20°C in a refrigerator for 60 min. After
centrifuging at 10,800 × g for 30 min, the supernatant was
discarded, 50 μl 75% ethanol was added and further centrifugation
at 10,800 × g for 30 min was conducted. The supernatant was
discarded. After drying, 7 μl Hi-Di (Shanghai Top Zhuo
Biotechnology Co., Ltd., Shanghai, China) was added to the residue,
which was the loaded onto an ABI 3730 genetic Analyzer (Haimai Pu
Biotechnology Co., Ltd.) for sequencing.
Results
Clinical manifestations
The clinical manifestations of the nine patients
with PKD are shown in Table II,
and the familial pedigrees of five of the cases are shown in
Fig. 1. The proband (PKD 2) in
family 1 had the chief complaint of involuntary limb swinging when
starting to run that was automatically relieved after lasting for
~30 sec. The consciousness of the patient was clear at onset and
the seizures occurred 3–10 times per day. The proband’s mother (PKD
1) reported dance-like movements of the upper limbs at the age of
10 years; however, the symptoms were mild and improved without
treatment. Questioning revealed that the proband’s grandfather had
a history of dance-like movements of the upper limbs. The proband
(PKD 4) in family 2 involuntarily stood from a seated position with
a twisted body posture and strange facial expressions, and the
father (PKD 3) had similar attacks at the ages of 10–18 years but
no longer experienced symptoms. Sporadic case PKD 5 had double-leg
spasms in sudden movements, each episode lasting for ~10 sec. PKD 6
and PKD 9 had lower limb stiffness in a sudden movement or tensing,
which was sustained for 5–15 sec; PKD 9 sometimes fell down. PKD 7
experienced involuntary limb swinging in sudden movements. PKD 8
had left upper limb spasticity in sudden movements or when changing
position.
| Table IIClinical data of PRRT2 gene test
results for nine patients with paroxysmal kinesigenic dyskinesia
(PKD). |
Table II
Clinical data of PRRT2 gene test
results for nine patients with paroxysmal kinesigenic dyskinesia
(PKD).
| Number | Gender | Family history | Onset age (years
old) | Main symptoms | Seizure
frequency | Drug treatment | PRRT2 mutation |
|---|
| PKD 1 | Female | Familial | 10 | Upper limb dance-like
movements | No symptoms
currently | Untreated | c.649dupC |
| PKD 2 | Male | Familial | 9 | Involuntary limb
swinging | 3–10 times per
day | Carbamazepine | c.649dupC |
| PKD 3 | Male | Familial | 10 | Involuntary twisting
of the body | No symptoms
currently | Phenytoin sodium | c.649dupC |
| PKD 4 | Female | Familial | 8 | Involuntary twisting
of the body | 1–5 times per
day | Carbamazepine | c.649dupC |
| PKD 5 | Male | Sporadic | 11 | Lower limb
spasms | 1 time per 2–3
days | Carbamazepine | None |
| PKD 6 | Female | Sporadic | 12 | Lower limb
stiffness | 1–3 times per
day | Sodium valproate | None |
| PKD 7 | Male | Sporadic | 5 | Involuntary limb
swinging | 3–5 times per
day | Carbamazepine | c.133-136delCCAG |
| PKD 8 | Male | Sporadic | 10 | Left upper limb
spasm | 5–20 times per
day | Carbamazepine | None |
| PKD 9 | Male | Sporadic | 12 | Lower limb stiffness
and falling down | 1–5 times per
day | Oxcarbazepine | None |
PRRT2 gene sequencing
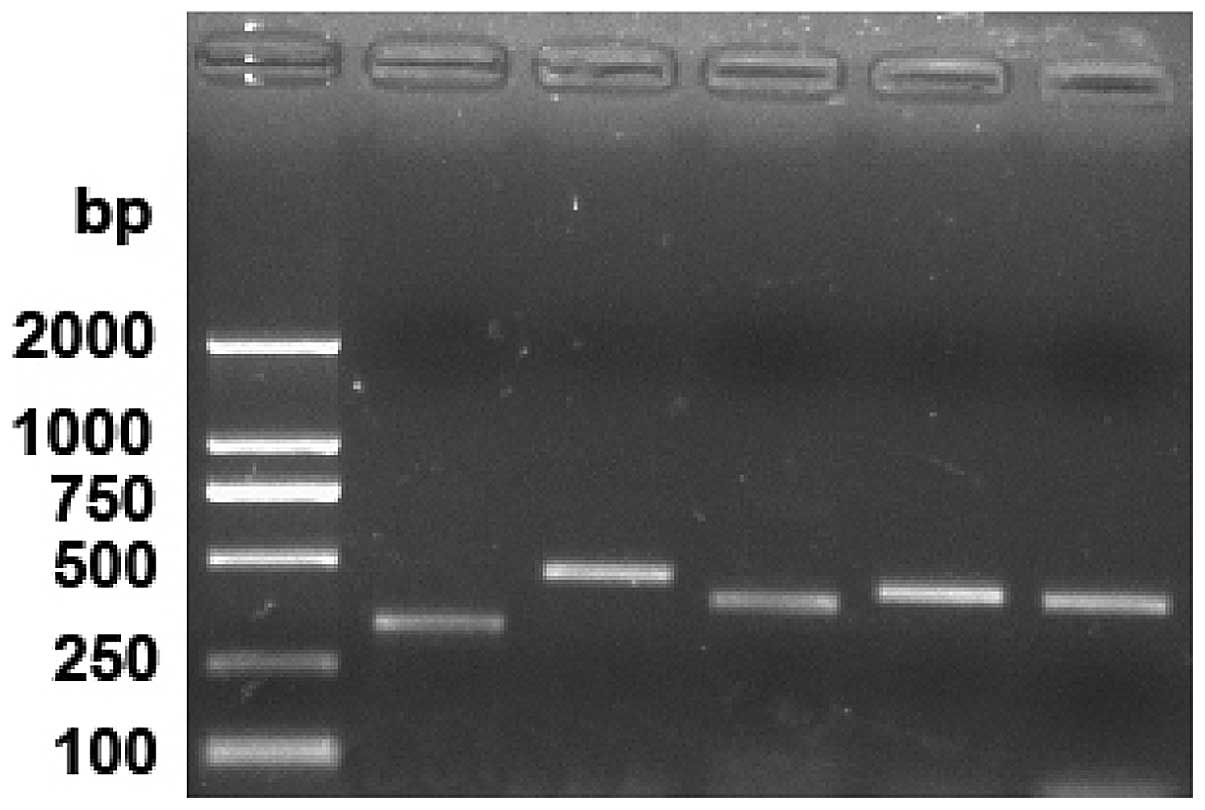
A gel map following PCR amplification is shown in
Fig. 2. The amplification bands of
the five PCR primers were 329, 467, 398, 433 and 431 bp
respectively.
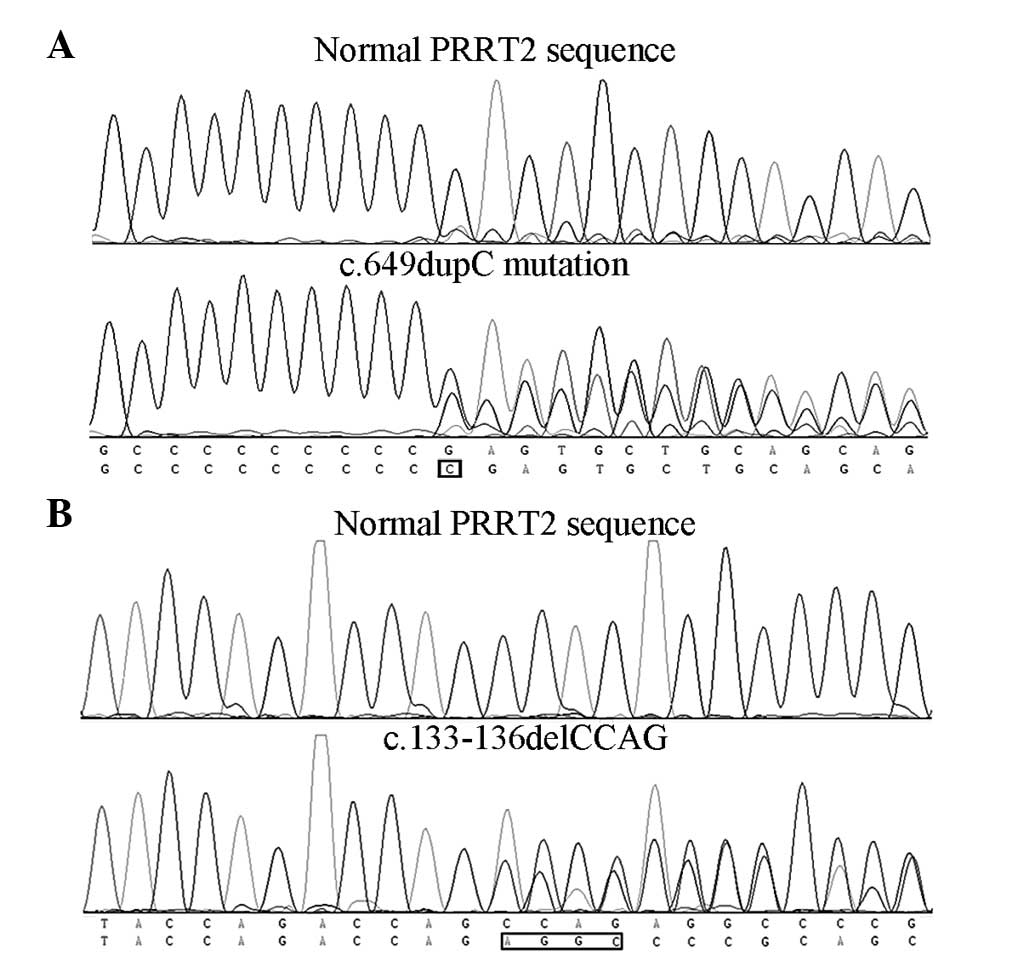
Sanger sequencing results showed that PKD 1–4
carried the PRRT2 gene c.649dupC (p.Arg217Profs*8) mutation
(Fig. 3A) and PKD 7 carried the
c.133-136delCCAG (p.Pro45Argfs*44) mutation (Fig. 3B), while no PRRT2 gene mutations
were identified in the remaining patients. Since PKD 7 was a
sporadic case, further screening for the mutation was performed and
it was found that the asymptomatic father also carried the
mutation.
Discussion
PKD is the most common form of paroxysmal dyskinesia
with onset triggered by sudden movements. Since PKD was first
reported in 1967, a number of academics and clinicians have been
committed to identifying the genes that cause PKD. In 2011, Chen
et al reported internationally that PRRT2 was a PKD
disease-associated gene (9), and
this was soon confirmed by other research groups (10–12).
It has now been confirmed that PRRT2 is a causative gene of PKD.
The findings of Chen et al provided the foundation for the
clinical molecular diagnosis of PKD.
However, few studies have reported on PKD. Song and
Hu described in detail the clinical manifestations of five PKD
cases (13); Li et al
reported seven PKD patients (14)
and Lin et al reported three PKD pedigrees with clinical and
genetic characteristics (15). Mao
et al reported the clinical features of 34 PKD patients
(16). However, PKD cases
confirmed by genetic methods were not reported. In the present
study, nine cases with a clinical diagnosis of PKD were collected,
including four cases with a family history. It was found that all
four cases with familial PKD and one case with sporadic PKD carried
PRRT2 mutations by Sanger sequencing of the PRRT2 gene, which
further confirmed that PRRT2 is a causative gene of PKD. In family
5, the proband PKD 7 carried a PRRT2 gene mutation at the
c.133-136delCCAG site; however, further testing found that the
asymptomatic father of PKD 7 also carried the same genetic
mutation. This suggests that PRRT2 mutations have incomplete
penetrance, that is, that certain individuals carrying the gene
mutations of PKD do not exhibit clinical symptoms. The reason for
the incomplete penetrance of PRRT2 gene mutations may be influenced
by the environment or other genetic modifications.
Currently, >30 PRRT2 mutations have been
identified in patients with PKD (9–12,17–21),
of which >95% are truncated mutants, while only ~5% are missense
mutations and splicing mutations (22). In the PRRT2 gene mutations,
c.649dupC is the most common site, accounting for ~80% of cases
(19). The high incidence of the
mutations has prompted c.649dupC to be defined as a high-frequency
mutation. In the present study, four of the five patients carrying
PRRT2 mutations had c.649dupC mutations, accounting for 80%, which
confirms the high frequency of this mutation. The study by Li et
al found that c.649dupC mutations can be de novo
mutations (23), suggesting that
the mutation was not from a founder effect but was a hotspot
mutation. Other studies support that c.649dupC is a hotspot
mutation (18,19).
Although PRRT2 genes of PKD have been cloned, the
function of PRRT2 remains unclear. Co-immunoprecipitation results
suggest that PRRT2 interacts with SNAP25 (12,24).
SNAP25 is a presynaptic membrane protein involved in the docking
and fusion of synaptic vesicles and associated with the release of
neurotransmitters (25). Since
SNAP25 forms a SNARE complex with syntaxin and synaptobrevin
(26), this suggests that PRRT2
may have functional connectivity with the SNARE complex, or may be
a component of the SNARE complex. In addition, SNAP25 induces
synaptic vesicles to release neurotransmitter when triggered by
Ca2+, indicating that PRRT2 mutations are likely to
cause abnormalities in neurotransmitter release (27). Whether PRRT2 mutations affect
intracellular Ca2+ influx remains unclear, and requires
further investigation.
The present study confirmed that PRRT2 is a
pathogenic gene of PKD, and that c.649dupC is a high frequency
mutation site. In addition, the presence of incomplete penetrance
of PRRT2 was revealed.
References
|
1
|
Bennett LB, Roach ES and Bowcock AM: A
locus for paroxysmal kinesigenic dyskinesia maps to human
chromosome 16. Neurology. 54:125–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Demirkiran M and Jankovic J: Paroxysmal
dyskinesias: clinical features and classification. Ann Neurol.
38:571–579. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhatia KP: Paroxysmal dyskinesias. Mov
Disord. 26:1157–1165. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruno MK, Hallett M, Gwinn-Hardy K, et al:
Clinical evaluation of idiopathic paroxysmal kinesigenic
dyskinesia: new diagnostic criteria. Neurology. 63:2280–2287. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kertesz A: Paroxysmal kinesigenic
choreoathetosis. An entity within the paroxysmal choreoathetosis
syndrome Description of 10 cases, including 1 autopsied. Neurology.
17:680–690. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tomita H, Nagamitsu S, Wakui K, et al:
Paroxysmal kinesigenic choreoathetosis locus maps to chromosome
16p11.2-q12.1. Am J Hum Genet. 65:1688–1697. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valente EM, Spacey SD, Wali GM, et al: A
second paroxysmal kinesigenic choreoathetosis locus (EKD2) mapping
on 16q13-q22.1 indicates a family of genes which give rise to
paroxysmal disorders on human chromosome 16. Brain. 123:2040–2045.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spacey SD, Valente EM, Wali GM, et al:
Genetic and clinical heterogeneity in paroxysmal kinesigenic
dyskinesia: evidence for a third EKD gene. Mov Disord. 17:717–725.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen WJ, Lin Y, Xiong ZQ, et al: Exome
sequencing identifies truncating mutations in PRRT2 that cause
paroxysmal kinesigenic dyskinesia. Nat Genet. 43:1252–1255. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Zhu X, Wang X, et al: Targeted
genomic sequencing identifies PRRT2 mutations as a cause of
paroxysmal kinesigenic choreoathetosis. J Med Genet. 49:76–78.
2012. View Article : Google Scholar :
|
|
11
|
Wang JL, Cao L, Li XH, et al:
Identification of PRRT2 as the causative gene of paroxysmal
kinesigenic dyskinesias. Brain. 134:3493–3501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee HY, Huang Y, Bruneau N, et al:
Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia
with infantile convulsions. Cell Rep. 1:2–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song F and Hu ZY: Clinical features and
pathogenesis of paroxysmal dyskinesias. J Clin Neurol (China).
18:382–384. 2005.(In Chinese).
|
|
14
|
Li SY, Liu WP, Xiong K and Xiao B:
Paroxysmal exercise-induced dyskinesia (clinical report of 7
cases). Stroke Nerv Dis. 13:48–49. 2006.(In Chinese).
|
|
15
|
Lin Y, Wu ZY, Wang N and Murong SX: The
clinical and genetic features of familial paroxysmal kinesigenic
dyskinesia:the three families reports. Zhonghua Shen Jing Ge Za
Zhi. 39:734–737. 2006.(In Chinese).
|
|
16
|
Mao CY, Shi CH, Song B, et al:
Genotype-phenotype correlation in a cohort of paroxysmal
kinesigenic dyskinesia cases. J Neurol Sci. 340:91–93. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Méneret A, Grabli D, Depienne C, et al:
PRRT2 mutations: a major cause of paroxysmal kinesigenic dyskinesia
in the European population. Neurology. 79:170–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao L, Huang XJ, Zheng L, Xiao Q, Wang XJ
and Chen SD: Identification of a novel PRRT2 mutation in patients
with paroxysmal kinesigenic dyskinesias and c.649dupC as a mutation
hot-spot. Parkinsonism Relat Disord. 18:704–706. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee YC, Lee MJ, Yu HY, et al: PRRT2
mutations in paroxysmal kinesigenic dyskinesia with infantile
convulsions in a Taiwanese cohort. PLoS One. 7:e385432012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Vliet R, Breedveld G, de Rijk-van,
Andel J, et al: PRRT2 phenotypes and penetrance of paroxysmal
kinesigenic dyskinesia and infantile convulsions. Neurology.
79:777–784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li HF and Wu ZY: PRRT2 mutations and PRRT2
disorders. Human Genet Embryol. 3:1052013.
|
|
22
|
Heron SE and Dibbens LM: Role of PRRT2 in
common paroxysmal neurological disorders: a gene with remarkable
pleiotropy. J Med Genet. 50:133–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li HF, Ni W, Xiong ZQ, Xu J and Wu ZY:
PRRT2 c.649dupC mutation derived from de novo in paroxysmal
kinesigenic dyskinesia. CNS Neurosci Ther. 19:61–65. 2013.
View Article : Google Scholar
|
|
24
|
Stelzl U, Worm U, Lalowski M, et al: A
human protein-protein interaction network: a resource for
annotating the proteome. Cell. 122:957–968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jarvis SE and Zamponi GW: Masters or
slaves? Vesicle release machinery and the regulation of presynaptic
calcium channels. Cell Calcium. 37:483–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sørensen JB, Matti U, Wei SH, et al: The
SNARE protein SNAP-25 is linked to fast calcium triggering of
exocytosis. Proc Natl Acad Sci USA. 99:1627–1632. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Prescott GR, Gorleku OA, Greaves J and
Chamberlain LH: Palmitoylation of the synaptic vesicle fusion
machinery. J Neurochem. 110:1135–1149. 2009. View Article : Google Scholar : PubMed/NCBI
|