Introduction
Cardiovascular disease is a major disease that
threatens human health, with the main pathological feature being
atherosclerosis (AS). The identification of methods for suppressing
intimal hyperplasia and delaying the progress of AS remains a
worldwide challenge (1). There is
currently no effective preventive measure for diseases with intimal
plaque hyperplasia as the main pathological feature; in particular,
there is no drug that directly targets the molecular components of
blood-intimal atherosclerotic plaques.
Rapamycin and its derivatives can inhibit the
proliferation of vascular smooth muscles (2). In certain research, rapamycin
exhibited significant effects against restenosis occurring
post-coronary intervention when used as a drug-coated stent
(3). While a drug-coated stent is
effective in the focal vessel, it is not able to act at non-stent
covered sites or in other systemic diseases, such as AS, which
cannot be treated with a stent (4). Moreover, rapamycin is a drug with
poor solubility that is unstable in stomach acid, and has an oral
bioavailability of only 14%. Therefore, it cannot play a systemic
role through oral or intravenous administration methods (5).
In the present study, rapamycin was encapsulated
into liposomes. In the future, these may be modified with an
antibody or receptor, so that the liposomes are actively targeted
via antigen-antibody or substrate-receptor combination towards the
molecular components of atherosclerotic plaques, thus developing a
receptor- or antibody-mediated active targeting administration
system towards atherosclerotic plaques. Such a system could greatly
increase the concentration of drug at the focal site, and provide a
new prevention method with active targeting towards a variety of
diseases involving AS.
In this study, the ethanol injection method was used
to prepare a liposomal rapamycin-delivery system, and the
formulation was optimized. Reverse dialysis was used to investigate
the in vitro release characteristics of the
rapamycin-containing liposomes, with the aim of exploring the
mechanism, method and characteristics of in vitro release.
The findings should lay a foundation for the further development of
a liposomal delivery system for rapamycin, actively targeted
towards the cellular components of atherosclerotic plaques.
Materials and methods
Preparation of rapamycin-containing
liposomes
Various amounts of phospholipids (Shanghai Taiwei
Pharmaceutical Co., Ltd., Shanghai, China), cholesterol (Tianjin
Bodi Chemical Co., Ltd., Tianjin, China) and rapamycin (purity,
99.2%; Shanghai Qi Ao Chemical Science Co., Ltd., Shanghai, China)
were dissolved in various amounts of absolute ethanol. Then, under
stirring, the above mixture was slowly and uniformly injected into
phosphate buffer (Tianjin Bodi Chemical Co., Ltd.), and the
resulting mixture was stirred at 60°C. The ethanol was then removed
by reduced-pressure evaporation, and the obtained crude liposome
solution was then sequentially filtered through 0.8-, 0.45-, 0.22-
and 0.1-μm membranes, five times each, for particle preparation.
Finally, rapamycin-containing liposomes were obtained.
Establishment of the method for the
determination of rapamycin content
This was conducted using a Diamonsil C18 column
(150×4.6 mm, 5 μm; Dikma Technologies Inc., Lake Forest, CA, USA)
with Hitachi Pump L-2130 and UV Detector L-2400 (Hitachi, Tokyo,
Japan). The mobile phase was methanol (chromatographic grade;
Jiangsu Hanbang Technology Co., Ltd., Huai’an, China) and water
(78:22), with a flow rate of 1.0 ml/min. The UV detection
wavelength was 278 nm, the column temperature was 50°C and the
injection volume was 20 μl
A 10.0 mg sample of rapamycin was precisely weighed
into a 100-ml volumetric flask, dissolved in acetonitrile
(chromatographic grade; Jiangsu Hanbang Technology Co., Ltd.) and
diluted to the 100 ml mark, which gave a stock solution of
rapamycin at the concentration of 100 mg/l. A precise amount of
this stock solution was diluted with acetonitrile to form solutions
with concentrations of 2.0, 5.0, 10.0, 20.0, 50.0 and 100.0 mg/l.
The samples were analyzed according to the chromatographic
conditions described above. Then, a linear regression plot was
prepared of mass concentration (C) to peak area (A). The standard
curve had the formula: A = 55307C − 9873.2 (r=1), indicating that
the linear relationship of rapamycin was good in the range of 2 to
100 mg/l.
Into a 10-ml flask was added 0.5 ml blank liposome,
followed by 0.5, 2.0 or 5.0 ml stock rapamycin solution. Methanol
was used to break the liposomes and for dilution. The process was
repeated three times, and sample solutions with concentrations of
5.0, 20.0 and 50.0 mg/l were obtained for injection and the
calculation of recovery. The recovery rates were 99.73, 100.5 and
98.02%, respectively, with relative standard deviations of 0.24,
1.70 and 1.74 %, respectively.
Determination of encapsulation efficiency
(EE)
Sephadex G-50 microcolumn-centrifuging
high-performance liquid chromatography (HPLC) was used to separate
the liposomes and free rapamycin for the determination of EE
(6,7). The specific steps were as follows:
Sephadex G-50 (Bio-Rad, Hercules, CA, USA), which was fully swelled
with distilled water, was placed into a 5-ml syringe, then
centrifuged at 600 × g for 3 min, dehydrated and used to establish
the Sephadex G-50 microcolumn. Following the addition of 0.5 ml
rapamycin-containing liposomes to the top of the microcolumn, the
column was centrifuged at 600 × g for 3 min and the separated
liquid was collected. Then, 0.5 ml pH 7.4 buffer was continuously
added and the column was centrifuged using the same method to elute
the liposomes. The above process was repeated twice, and the eluent
was collected. Methanol was used to break the liposomes and dilute
to a volume of 10 ml prior to sample determination to calculate the
concentration of rapamycin (C1) encapsulated inside the
liposomes. Another 0.5 ml liposomes were directly diluted with
methanol to the same extent, but were not subjected to microcolumn
centrifugation, and sample determination was conducted to calculate
the rapamycin concentration C2. EE was then calculated
using the formula: EE (%) = C1/C2 × 100.
Statistical methods
Experimental data were statistically processed using
SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA) and were
expressed as mean ± standard deviation. The results of orthogonal
experiments were analyzed by multivariate analysis of variance
(MANOVA), while partial results were analyzed by ANOVA.
Results
Investigation of univariate factors of
the rapamycin-containing liposome formulation
The phospholipid-cholesterol mass ratio, the
drug-lipid ratio and the aqueous phase pH of the formulation were
fixed; only the concentrations of phospholipid was changed to
prepare liposomes with phospholipid concentrations of 1, 2, 3, 4
and 5%, with the aim of investigating the impact of phospholipid
concentrations on the EE. The results demonstrated that in these
experimental conditions, as the phospholipid concentration
increased, the EE increased. However, excessively high phospholipid
concentrations resulted in the aggregation of phospholipids, making
it difficult for the rapamycin to be released.
The phospholipid concentration, drug-lipid ratio and
the aqueous phase pH of the formulation were fixed; only the
quantity of cholesterol was changed to prepare liposomes with
phospholipid-cholesterol mass ratios of 15:1, 10:1, 8:1, 6:1, 4:1
and 3:1. The results indicated that the EE exhibited a trend of
increasing initially and then decreasing as the amount of
cholesterol incorporated into the liposomes increased. Cholesterol
played the role of a regulating agent towards liposome membrane
fluidity, and improved the drug EE and stability. However,
excessive amounts of cholesterol competed for position in the
phospholipid bilayer position with the liposoluble drug, namely
rapamycin, resulting in decreased EE.
The phospholipid concentration,
phospholipid-cholesterol mass ratio and the aqueous phase pH of the
formulation were fixed, and the drug-lipid ratio was established at
1:10, 1:15, 1:20, 1:30 and 1:40 to prepare the liposomes. The
results indicated that when the drug-lipid ratio was greater than
1:20, the EE was low.
The phospholipid concentration,
phospholipid-cholesterol mass ratio and drug-lipid ratio of the
formulation were fixed, and the ethanolic solution of the
formulation was injected into phosphate buffer with a pH of 5.8,
6.5, 7.0, 7.4 or 8.0 to prepare liposomes. The results demonstrated
that liposomes prepared at a pH of between 6.5 and 7.4 were stable
and the EE was high, while flocculation or aggregation occurred
under other pH conditions.
Optimization of the rapamycin-containing
liposome formulation by orthogonal design
Based on the investigation of single factors, the
four factors, namely phospholipid concentration (factor A),
phospholipid-cholesterol mass ratio (factor B), drug-lipid ratio
(factor C) and aqueous phase pH (factor D) were selected, and three
levels of each factor were designated for the orthogonal design,
with the EE as the investigating indicator to screen the
formulation. The orthogonal factors and levels are shown in
Table I.
 | Table IFactors and levels. |
Table I
Factors and levels.
| Factors |
|---|
|
|
|---|
| Levels | A | B | C | D |
|---|
| Level 1 | 2 | 10:1 | 1:15 | 7.4 |
| Level 2 | 3 | 8:1 | 1:20 | 7.0 |
| Level 3 | 4 | 6:1 | 1:30 | 6.5 |
The L9 (34) orthogonal table
(8) was used and nine formulations
were obtained, according to the above designs. Rapamycin-containing
liposomes were prepared for EE determination. The orthogonal test
results are shown in Table II,
and the variance analysis is shown in Table III.
| Table IIResults of orthogonal testing. |
Table II
Results of orthogonal testing.
| No. | A | B | C | D | EE % |
|---|
| 1 | 1 | 1 | 1 | 1 | 71.75 |
| 2 | 1 | 2 | 2 | 2 | 74.64 |
| 3 | 1 | 3 | 3 | 3 | 72.80 |
| 4 | 2 | 1 | 2 | 3 | 75.61 |
| 5 | 2 | 2 | 3 | 1 | 81.16 |
| 6 | 2 | 3 | 1 | 2 | 70.49 |
| 7 | 3 | 1 | 3 | 2 | 85.64 |
| 8 | 3 | 2 | 1 | 3 | 77.39 |
| 9 | 3 | 3 | 2 | 1 | 84.28 |
| K1 | 219.19 | 233.00 | 219.63 | 237.19 | |
| K2 | 227.26 | 233.19 | 234.53 | 230.77 | |
| K3 | 247.31 | 227.57 | 239.60 | 225.80 | |
| K̄1 | 73.06 | 77.67 | 73.21 | 79.06 | |
| K̄2 | 75.75 | 77.73 | 78.18 | 76.92 | |
| K̄3 | 82.44 | 75.86 | 79.87 | 75.27 | |
| R | 9.38 | 1.87 | 6.66 | 3.79 | |
| Table IIIVariance analysis. |
Table III
Variance analysis.
| Source | S | f | MS | F-statistic | P-value |
|---|
| Factor A | 139.98 | 2 | 69.99 | 635.27 | <0.01 |
| Factor B | 6.78 | 2 | 3.39 | 30.82 | <0.05 |
| Factor C | 71.92 | 2 | 35.96 | 326.91 | <0.01 |
| Factor D | 21.67 | 2 | 10.84 | 98.55 | <0.01 |
| Error | 0.22 | 2 | 0.11 | | |
It can be determined by direct-viewing analysis of
the extremum values (R) in Table
II that the importance degrees of the factors were in the
order: A>C>D>B, and the optimized formulation composition
was A3B2C3D1, that is,
the phospholipid concentration was 4%, the phospholipid-cholesterol
ratio was 8:1, the drug-lipid ratio was 1:30 and the pH was
7.4.
The variance analysis indicates that factors A, C
and D had significant impacts on the experimental results, and
factor B was also significant. According to the F value, the
impacts of various factors towards the test results were
A>C>D>B, which is consistent with the results of
direct-viewing analysis.
The optimized formulation had a drug-lipid ratio of
1:30; however, considering the amount of liposomal drug loading, a
drug-lipid ratio of 1:20 was selected. As for factor C, the levels
2 and 3 were subjected to single-factor variance analysis, and the
result was F<F0.05 (1,4)=7.71, indicating that the levels 2 and
3 of factor C were not statistically significant towards the
experimental results. The final formulation was adjusted to
A3B2C2D1, that is, the
phospholipid concentration was 4%, the phospholipid-cholesterol
ratio was 8:1, the drug-lipid ratio was 1:20 and the pH value was
7.4. The preparation and analysis of the above optimal formulation
was repeated three times, and the EE of the rapamycin-containing
liposomes was measured as 82.11±2.13%.
Investigation of in vitro
release
A previously described reverse dialysis method
(9) was modified for the
determination of the in vitro release of rapamycin from the
liposomes.
In the reverse dialysis, 500 ml 20% ethanol was used
as the release medium; 10 ml release medium was drawn and placed
into a dialysis bag (diameter, 25 mm; trapping substances with a
relative molecular mass of 12,000–14,000; Beijing Huamei
Biotechnology Co., Ltd, Beijing, China). The dialysis bag was then
clamped and attached on the paddle of a dissolution apparatus; 5 ml
rapamycin-containing liposomes and 5 ml ethanol solution of
rapamycin with the same drug content as the liposomes, were
respectively dissolved in the dialysis vessel, with stirring at
37°C and 300 × g. Sampling of 100 μl liquid from the dialysis bag
was conducted at 1, 2, 4, 6, 8, 10, 12 and 24 h for sample
determination and calculation of the accumulative release rate. A
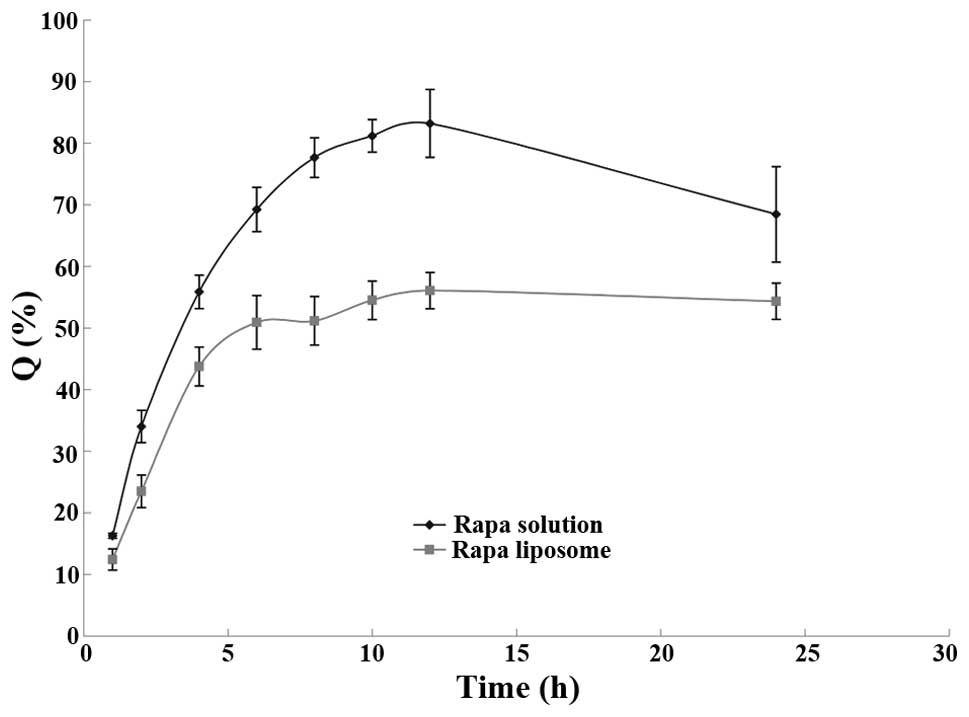
release curve was drawn using time (t) as the abscissa, and the
accumulative release rate (Q%) as the ordinate; the resulting curve
is shown in Fig. 1.
It was found from the experiment that after 12 h,
the amount of drug released gradually decreased with the extension
of release time. Theoretically, the accumulative release of the
drug should be maintained at a high level; however, at 37°C,
rapamycin was unstable in aqueous solution and the drug content was
reduced (10). Thus, it was
necessary to consider the degradation of rapamycin in the release
medium.
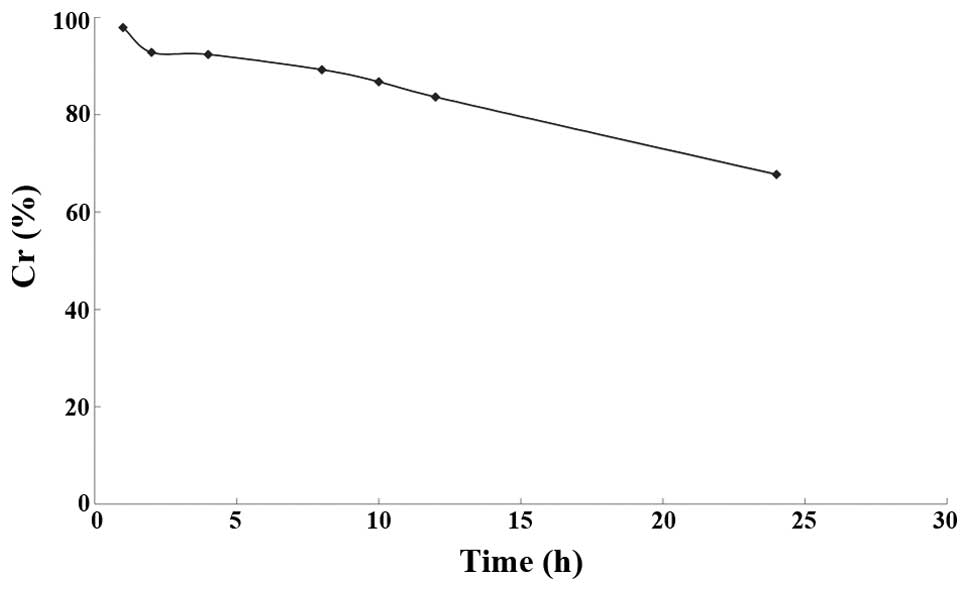
The 20% ethanolic solution of rapamycin was placed
in a 37°C water bath, and sampled at 0, 1, 2, 4, 8, 10, 12 and 24
h, respectively, for the determination of rapamycin concentration
(Ct) by HPLC at different time points. The ratio Ct/Co, where Co is
the rapamycin concentration at 0 h, was used to calculate the
residual percentage Cr, with the time t as the abscissa and Cr as
the ordinate. The resulting curve is shown in Fig. 2.
The results demonstrated that rapamycin was unstable
in the release medium; after 24 h, the drug content was 67.72% of
the initial content. Linear fitting plots for zero-, first- and
second-order models were drawn to study the degradation kinetics
(Table IV). The linear fitting
results revealed that the degradation kinetics of rapamycin fitted
the first-order model.
| Table IVDegradation curve-fitting equations
of rapamycin solution in release medium. |
Table IV
Degradation curve-fitting equations
of rapamycin solution in release medium.
| Model | Fitting
equation | Correlation
coefficient r |
|---|
| Zero-order
model | Cr = −0.0122 t +
0.9397 | 0.9786 |
| First order
model | lnCr = −0.0162 t +
0.0095 | 0.9931 |
| Second order
model | 1/Cr = 0.0202 t +
0.9711 | 0.9809 |
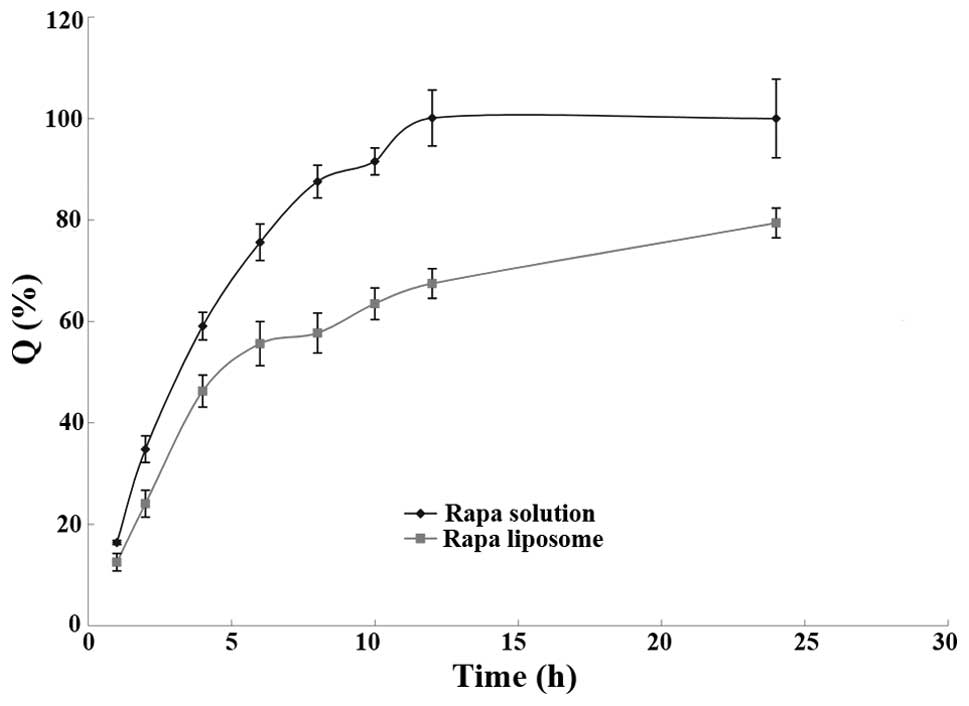
The amount of released drug at each time point was
entered into the fitted degradation kinetics equation to calculate
the amount of degraded drug; the accumulation of the released and
degraded drug was set as the ordinate and the time t was set as the
abscissa to fit the release profile of the rapamycin-containing
liposomes. The resulting curve is shown in Fig. 3.
As can be observed in Fig. 3, the liposomes had a
sustained-release effect for rapamycin; the 24 h total release dose
was 80%. The release process of rapamycin from the liposomes can be
divided into two phases: In the first 4 h, the release rate was
fast and ~50% of the total dose was released; this phase was the
rapid release phase. This may be due to the release of the
encapsulated free drug and the drug that was adsorbed on the
surface of the liposomes by a weak binding force. Four hours later,
the drug release became relatively slow, known as the slow release
phase. The rapamycin solution and rapamycin-containing liposomes
were fitted according to the release models including the Higuchi
(11) Ritger-Peppas (12) and Weibull (13) models; the results are shown in
Tables V and VI. The results indicated that the in
vitro release profile of rapamycin from the liposomes and
solution best fitted the first-order release model.
| Table VRelease curve-fitting equations of
rapamycin solution. |
Table V
Release curve-fitting equations of
rapamycin solution.
| Model | Fitting
equation | Correlation
coefficient r |
|---|
| Zero-order release
model | Q = 0.073 t +
0.2158 | 0.9181 |
| First-order release
model | ln(1-Q) = −0.2609 t
+ 0.1046 | 0.9959 |
| Higuchi model | Q = 0.3402
t1/2 − 0.1289 | 0.9802 |
| Ritger-Peppas
model | lnQ = 0.7144 lnt −
1.6521 | 0.9076 |
| Weibull model | ln(1/1-Q) = 0.9816
lnt − 0.1067 | 0.9003 |
| Table VIRelease curve-fitting equations of
rapamycin-containing liposomes. |
Table VI
Release curve-fitting equations of
rapamycin-containing liposomes.
| Model | Fitting
equation | Correlation
coefficient r |
|---|
| Zero-order release
model | Q = 0.0152 t +
0.4544 | 0.9101 |
| First-order release
model | ln(1-Q) = −0.046 t
− 0.5090 | 0.9770 |
| Higuchi model | Q = 0.1166
t1/2 + 0.2697 | 0.9673 |
| Ritger-Peppas
model | lnQ = 0.2965 lnt −
0.1067 | 0.9600 |
| Weibull model | ln(1/1-Q) = 0.5311
lnt − 0.1704 | 0.9710 |
Discussion
With the use of new technology and equipment, and
the development and application of excellent carrier materials and
accessories, targeted drug delivery (TDD) technology has been
developing rapidly in recent years, and has gradually extended to
the treatment fields of multiple diseases (14–17).
Although nanoparticles, nanocapsules, microspheres,
microcapsule, micelle multimers and monoclonal antibody coupling
may be considered as the ideal medicament carriers, liposomes
remain inexpensive, readily available and more studied drug
carriers (18–21).
Liposomes are extensively researched TDD carriers,
with a sustained-release, long-lasting effect, low systemic
toxicity and good biocompatibility, and are suitable for
administration by a variety of routes. However, traditional
liposomes only have a passive targeting role; if antibody or ligand
molecules, which are targeted towards proliferated intimal tissues,
are connected to the liposomal membrane and thus form targeting
liposomes, active targeting can be achieved, significantly
enhancing the concentration of liposomes in vascular tissues,
particularly in atherosclerotic plaques (22–26).
The liposomal bilayer is amenable to surface modification to
achieve TDD towards the cardiovascular system.
Rapamycin is a lipophilic substance, which is easily
compatible with the hydrophobic chains that are used in the
preparation of liposomes, and form a part of the bilayer. The
optimum mass ratio of phospholipids, cholesterol and rapamycin is
that at which liposomes with the highest EE are formed. In this
study, the ethanol injection method was used to prepare
rapamycin-containing liposomes, a microcolumn centrifugation HPLC
method was used to determine the EE, and the EE was used as the
evaluation indicator to inspect the impacts of phospholipid
concentration, phospholipid-cholesterol mass ratio, drug-lipid
ratio and aqueous phase pH on the liposomes. Based on these
evaluations, an orthogonal design experiment was performed to
optimize the formulation. The orthogonal test results revealed that
the best formulation had a phospholipid concentration of 4%,
phospholipid-cholesterol mass ratio of 8:1, drug-phospholipid mass
ratio of 1:20 and aqueous phase pH of 7.4. The encapsulation rate
of the resultant rapamycin-containing liposomes was high, reaching
82.11±2.13%, and the reproducibility was good. The higher EE also
indicated that the lipid bilayer was able to significantly
solubilize the hydrophobic drug rapamycin, enabling it to be
administered through the inner vessel, which would be an effective
means to resolve the low oral bioavailability of rapamycin.
The EE is an important indicator when evaluating a
liposomal delivery system, and there are numerous methods for
determining it. In this study, Sephadex column chromatography was
initially used, with online elution using a buffer of pH 7.4;
however, free rapamycin did not dissolve in the buffer and could
not be eluted quickly. Subsequently, a dialysis method was used to
isolate the liposomes and free the drug. As rapamycin is strongly
liposoluble, the unencapsulated drug was present in the external
aqueous phase in the form of small crystals, which would not pass
through the dialysis bag; therefore, the measured quantity of free
drug was likely to be inaccurate. Finally, with reference to the
literature, a microcolumn centrifugation HPLC method was
established to determine the EE; the method was simple and
reproducible.
For drugs with poor solubility, when performing a
release characteristics study, a surfactant or an organic solvent
is typically used to improve the drug solubility in the release
medium to meet sink conditions. In the present study, ethanol was
selected as a cosolvent to increase the solubility of rapamycin.
When the rapamycin-containing liposomes reached sink conditions,
drug release was not complete in 24 h, indicating that the
liposomes had sustained release effects.
Currently, the main method used in in vitro
release studies of liposomal preparations is the dialysis method
(27). In this method, liposomes
contact only a small amount of release medium in the dialysis bag;
when the drug diffuses from the liposomes and is released into the
release medium in the bag, it then diffuses through the dialysis
bag to a massive release medium. As the drug concentration gradient
inside and outside the dialysis bag is small, passive diffusion is
slow, so that the release rate is low. In the present study, the
reverse dialysis method was used to determine the in vitro
release of rapamycin from the liposomes (9); since the liposomes directly contacted
the release medium and were largely diluted, it better simulated
the state that the drug achieves following intravenous injection.
The experimental results indicated that the dialysis bag had almost
no adsorption of rapamycin, and the rapamycin recovery rate was
99.21±1.12% (n=3).
In order to investigate the release properties of
the rapamycin-containing liposomes, a release equation was used for
fitting, and the optimal equation was determined by means of the
correlation coefficient r. The results revealed that the release of
rapamycin from the solution and liposomes best fitted the
first-order release model, in which the in vitro release may
be described by a concentration-dependent permeation release model.
Since the in vitro release conditions were not the same as
the in vivo blood environment, the in vitro release
experiment did not truly reflect the situation of in vivo
release. Further studies of the correlation of in vivo
absorption and in vitro release of rapamycin from the
liposomal delivery system are required.
References
|
1
|
de Jager SC and Kuiper J: Vaccination
strategies in atherosclerosis. Thromb Haemost. 106:796–803. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sabers CJ, Martin MM, Brunn GJ, Williams
JM, Dumont FJ, Wiederrecht G and Abraham RT: Isolation of a protein
target of the FKBP12-rapamycin complex in mammalian cells. J Biol
Chem. 270:815–822. 1994. View Article : Google Scholar
|
|
3
|
Cheng-Lai A and Frishman WH:
Sirolimus-eluting coronary stents: novel devices for the management
of coronary artery disease. Am J Ther. 11:218–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klugherz BD, Llanos G, Lieuallen W, et al:
Stent-based delivery of sirolimus for the prevention of resrenosis.
J Am Coll Cardiol. 35(Suppl 1): 58A2000.
|
|
5
|
Gallo R, Padurean A, et al: Inhibition of
intimal thickening after balloon angioplasty in porcine coronary
arteries by targeting regulators of the cell cycle. Circulation.
99:2164–2170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang JA, Anyarambhatla G, Ma L, Ugwu S,
Xuan T, Sardone T and Ahmad I: Development and characterization of
a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU)
formulation. Eur J Pharm Biopharm. 59:177–187. 2005. View Article : Google Scholar
|
|
7
|
Li C and Deng Y: A novel method for the
preparation of liposomes: freeze drying of monophase solutions. J
Pharm Sci. 93:1403–1414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu YP, Lasne C and Chouroulinkov I: Use of
an orthogonal design method to study two-stage chemical
carcinogenesis in BALB/3T3 cells. Carcinogenesis. 7:893–898. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levy MY and Benita S: Drug release from
submicronized O/W emulsion: a new in vitro kinetic evaluation
model. Int J Pharm. 66:291990. View Article : Google Scholar
|
|
10
|
Rouf MA, Bilensoy E, Vural I and Hıncal
AA: Determination of stability of sirolimus following exposure to
different conditions. Eur J Pharm Sci. 32(Suppl): S462007.
|
|
11
|
Higuchi WI, Tzeng CS, Chang SJ, Chiang HJ
and Liu CL: Estimation of cholesterol solubilization by a mixed
micelle binding model in aqueous
tauroursodeoxycholate:lecithin:cholesterol solutions. J Pharm Sci.
97:340–349. 2008. View Article : Google Scholar
|
|
12
|
Ravi PR, Ganga S and Saha RN: Design and
in vitro evaluation of zidovudine oral controlled release tablets
prepared using hydroxypropyl methylcellulose. Chem Pharm Bull
(Tokyo). 56:518–524. 2008. View Article : Google Scholar
|
|
13
|
Lionberger RA, Raw AS, Kim SH, Zhang X and
Yu LX: Use of partial AUC to demonstrate bioequivalence of Zolpidem
Tartrate Extended Release formulations. Pharm Res. 29:1110–1120.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Masaka T, Matsuda T, Li Y, et al:
Synthesis of VIP-lipopeptide using a new linker to modify
liposomes: Towards the development of a drug delivery system for
active targeting. Chem Pharm Bull (Tokyo). 61:1184–1187. 2013.
View Article : Google Scholar
|
|
15
|
Wang X, Lin Y, Zeng Y, Sun X, Gong T and
Zhang Z: Effects of mycophenolic acid-glucosamine conjugates on the
base of kidney targeted drug delivery. Int J Pharm. 456:223–234.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pehlivan SB: Nanotechnology-based drug
delivery systems for targeting, imaging and diagnosis of
neurodegenerative diseases. Pharm Res. 30:2499–2511. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arias JL: Liposomes in drug delivery: a
patent review (2007 - present). Expert Opin Ther Pat. 23:1399–1414.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murakami M, Cabral H, Matsumoto Y, et al:
Improving drug potency and efficacy by nanocarrier-mediated
subcellular targeting. Sci Transl Med. 3:64ra22011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Musacchio T and Torchilin VP: Recent
developments in lipid-based pharmaceutical nanocarriers. Front
Biosci (Landmark Ed). 16:1388–1412. 2011. View Article : Google Scholar
|
|
20
|
Roger M, Clavreul A, Venier-Julienne MC,
Passirani C, Montero-Menei C and Menei P: The potential of
combinations of drug-loaded nanoparticle systems and adult stem
cells for glioma therapy. Biomaterials. 32:2106–2116. 2011.
View Article : Google Scholar
|
|
21
|
Suo X, Deng Y and Hao A: Determination of
lauroyl-indapamide in rat whole blood by high-performance liquid
chromatography. J Chromatogr B Analyt Technol Biomed Life Sci.
819:191–196. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Torchilin VP, Narula J, Halpern E and Khaw
BA: Poly(ethylene glycol)-coated anti-cardiac myosin
immunoliposomes: factors influencing targeted accumulation in the
infarcted myocardlium. Biochem Biophys Acta. 1279:75–83. 1996.
View Article : Google Scholar
|
|
23
|
Chen Y, Deng YJ and Hao YL: Surface
modification of liposomes for cardiomyocytes targeting in vitro.
Pharmazie. 60:238–240. 2005.PubMed/NCBI
|
|
24
|
Chen Y, Deng YJ, Hao YL, Hao AJ, Zhong HJ
and Wang XM: Uptake of liposomes by cultured cardiomyocytes.
Pharmazie. 60:844–848. 2005.PubMed/NCBI
|
|
25
|
Nag OK and Awasthi V: Surface engineering
of liposomes for stealth behavior. Pharmaceutics. 25:542–569. 2013.
View Article : Google Scholar
|
|
26
|
Paszko E and Senge MO: Immunoliposomes.
Curr Med Chem. 19:5239–5277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen J and Burgess DJ: In vitro
dissolution testing strategies for nanoparticulate drug delivery
systems: recent developments and challenges. Drug Deliv Transl Res.
3:409–415. 2013. View Article : Google Scholar : PubMed/NCBI
|