Introduction
Achondroplasia (ACH) is a hereditary dwarfism caused
by a disturbance in the proliferation and differentiation of growth
plate chondrocytes, followed by an impairment in endochondral bone
growth. The incidence rate of ACH is ~1/15–40,000 live births
(1). In total, between 80 and 90% of
ACH cases are sporadic (2). Newborn
infants with ACH typically present with disproportionate shortening
of the limbs, a long and narrow trunk, a large head with frontal
bossing and midfacial hypoplasia. The hands are short and broad,
and frequently exhibit a three-pronged (trident) configuration.
Furthermore, numerous joints, with the exception of the elbow, are
hyperextensible (3). The disease
shows an autosomal dominant inheritance and is caused by mutations
in the gene encoding the transmembrane receptor, fibroblast growth
factor receptor 3 (FGFR3), which is an important regulator of
linear bone growth. FGFR3 is expressed in various tissues including
the cartilage, brain, kidneys and the intestines at different
stages of development. FGFR3 mutations generate deficient proteins
that affect chondrocyte proliferation and calcification, and hinder
cartilage growth and development, thereby resulting in an external
phenotype of ACH (4). The human
FGFR3 gene is located on chromosome 4q16.3. In total, >90% of
patients with ACH have a G1138A mutation in the transmembrane
domain of the FGFR3 gene (5,6). Research on ACH began later in China
than in Europe and the US. Currently, almost 60 clinical cases have
been reported around the country and there have been no studies, to
the best of our knowledge, on the incidence rate of ACH in China.
In the present study, the clinical characteristics of a Chinese
male child diagnosed with ACH were analyzed, and tests for the
FGFR3 gene mutation were performed on the patient and patient's
family.
Case report
Patient characteristics and clinical
observations
A four-year-old male was admitted to the First
Hospital of Lanzhou University (Lanzhou, China) with growth
retardation since the age of three years. The patient was an only
child, and was born at full term via vaginal delivery with a birth
weight of 3,900 g. Teething began between seven and eight months,
and the patient was walking at one year. After one year of age, it
was noted that the patient's growth and development were slow, and
that his height was lower compared with other children of a similar
age. The annual increase in height was <5 cm; however, the
weight and intelligence level were normal. A physical examination
at the time of admission revealed the following characteristics:
Body temperature, 36°C; pulse rate, 90 bpm; respiratory rate, 20
breaths/min; height, 85 cm; and weight, 16 kg. The patient had a
large head with a prominent forehead. In addition, there was
disproportionate shortening of the upper arms and legs, and the
patient had short hands with fingers that assumed a three-pronged
(trident) configuration.
Laboratory results were as follows: Serum calcium,
2.41 mmol/l (normal range, 2.10–2.80 mmol/l); serum phosphorus,
1.66 mmol/l (normal range, 0.97–1.60 mmol/l); intact parathyroid
hormone, 24.20 pg/ml (normal range, 14–72 pg/ml); 25-hydroxy
vitamin D, 72.6 nmol/l (normal range, 47.7–144 nmol/l);
osteocalcin, 38.8 ng/ml (normal range, 12.8–55 ng/ml);
bone-specific alkaline phosphatase, 89.7 ng/ml (normal range,
7.3–22.4 ng/ml); urine calcium, 1.30 mmol/24 h (normal range,
2.5–7.5 mmol/24 h); urine phosphorus, 13.75 mmol/24 h (normal
range, 23–48 mmol/24 h); thyroid-stimulating hormone (TSH), 1.26
µIU/ml (normal range, 0.55–4.78 µIU/ml); triiodothyronine
(T3), 1.69 ng/ml (normal range, 0.60–1.81 ng/ml);
thyroxine (T4), 10.0 µg/dl (normal range, 4.50–10.9
µg/dl); free T3, 4.19 pg/ml (normal range, 2.3–4.2
pg/ml); and free T4, 1.34 ng/dl (normal range, 0.89–1.76
ng/dl). Test for antibodies against the TSH receptor, thyroid
peroxidase and thyroglobulin were negative. In addition, the levels
of sex hormones, cortisol and adrenocorticotropic hormone (ACTH)
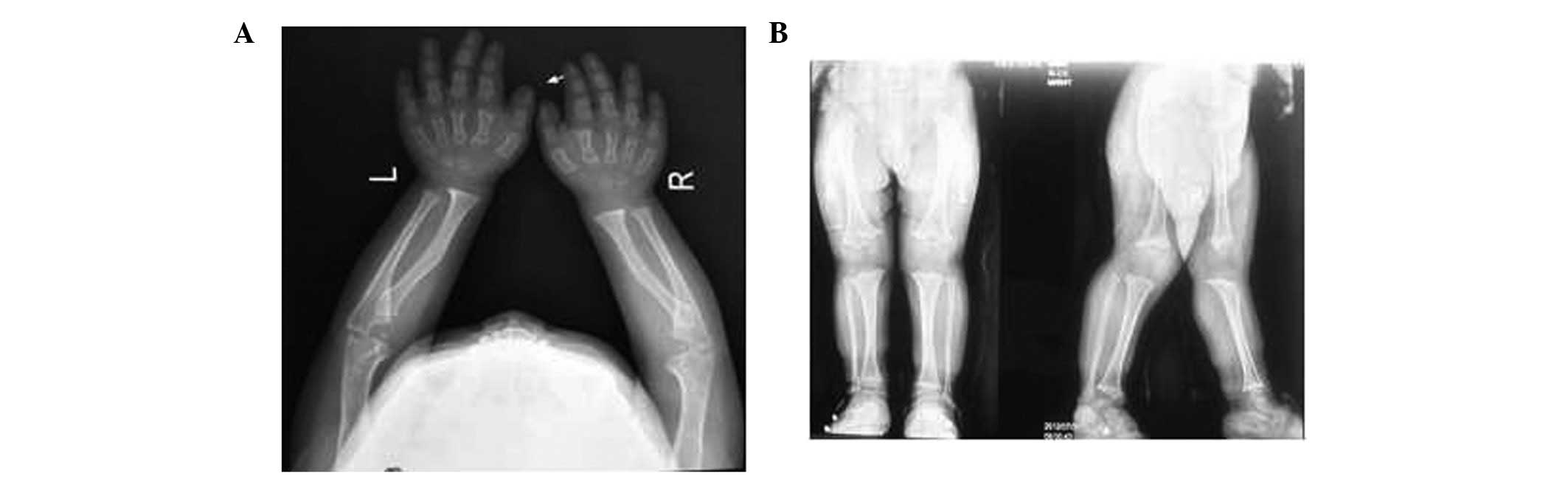
were normal. The patient's karyotype was 46,XY. A radiograph of the
upper limbs and hands revealed the hands to be short and broad,
with a trident configuration, and that the estimated age of the
wrist bones was lower than the actual age of the patient (Fig. 1A). A radiograph of the lower limbs
revealed disproportionate shortening of the limbs, bilateral
trumpet-like enlargement of the distal femoral metaphyses and
blurring of the epiphyseal shape (Fig.
1B). Furthermore, a magnetic resonance imaging (MRI) scan
revealed a decreased pituitary volume and a hyperintense and
spot-like corpus callosum, indicative of malacia, on the T1- and
T2-weighted images. No similar phenotype was identified in the
parents or other family members (grandfather, grandmother, aunts,
uncles and cousins) of the patient.
Growth hormone provocation tests
Growth hormone (GH) responses to provocation tests
(0.1 IU/kg insulin-induced hypoglycemia, 10 mg/kg L-dopa and 0.5
g/kg arginine) and the levels of GH during exercise (brisk walking
for 15 minutes followed by running for 5 min with the heart rate
reaching >120 beats/min; GH levels obtained 20 min after
exercise initiation) were evaluated. Serum GH concentrations were
determined using an immunoradiometric assay (Tianjin Nine Tripods
Medical & Bioengineering Co., Ltd., Tianjin, China). The
intra-assay variation was <5.8% and the inter-assay variation
was <9.3%. The responses of cortisol and ACTH to insulin-induced
hypoglycemia were evaluated simultaneously.
Peak GH levels of <10 ng/ml on GH provocation
tests with insulin, L-dopa and arginine, and a GH level of <5
ng/ml during exercise were defined as blunted GH secretion,
according to the criteria of the Foundation for Growth and Science
in China (7).
Genomic DNA extraction
A 2-ml sample of peripheral venous blood was
collected from the patient, his parents and seven healthy controls
(3 females and 4 males, aged 10–35 years) who were volunteers that
were in the hospital for a physical examination. DNA was isolated
from the blood samples using a Blood Genome DNA Extraction kit
(Takara Biotechnology Co., Ltd., Dalian, China), and subsequently
dissolved in Tris-EDTA buffer (Takara Biotechnology Co., Ltd.) and
stored at −20°C until required for further use.
DNA amplification and mutation
detection
Exon 10 of the FGFR3 gene was amplified using
polymerase chain reaction (PCR; Roche Diagnostics Co., Ltd.,
Shanghai, China). The sequence of the forward primer was
5-AGGCGCGTGCTGAGGTTCTGAG-3 and the sequence of the reverse primer
was 5-GGAGATCTTGTGCACGGTGG-3. (Sangon Biotech Co., Ltd., Shanghai,
China) All PCR amplifications were performed in a total volume of
50 µl, which contained 2 µl extracted DNA, 20 pmol each of the
forward and reverse primers, 19 µl 2X Taq PCR Master mix (Takara
Biotechnology Co., Ltd.) and 25 µl deionized water. Thermal cycling
conditions were as follows: Initial activation of DNA polymerase at
95°C for 5 min, followed by 30 cycles of 95°C for 30 min, 58°C for
30 min and 72°C for 45 min, and a final extension at 72°C for 5
min. The PCR products were purified by a UNIQ-10 Column kit (Sangon
Biotech Co., Ltd.) and sequenced directly on a ABI3730XL genetic
analyzer (Applied Biosystems Life Technologies, Foster City, CA,
USA) to detect the gene mutations. The sequence results were
contrasted with the normal sequence of the FGFR3 gene obtained from
GenBank. All mutations were confirmed through forward and reverse
sequencing.
Ethics statement
Written informed consent was obtained from the
patient's family. The study was approved by the Human Ethics Review
Committee of the First Hospital of Lanzhou University and complied
with the Declaration of Helsinki for Experimentation on Humans,
1975 (revised in 1983).
Results
GH provocation tests
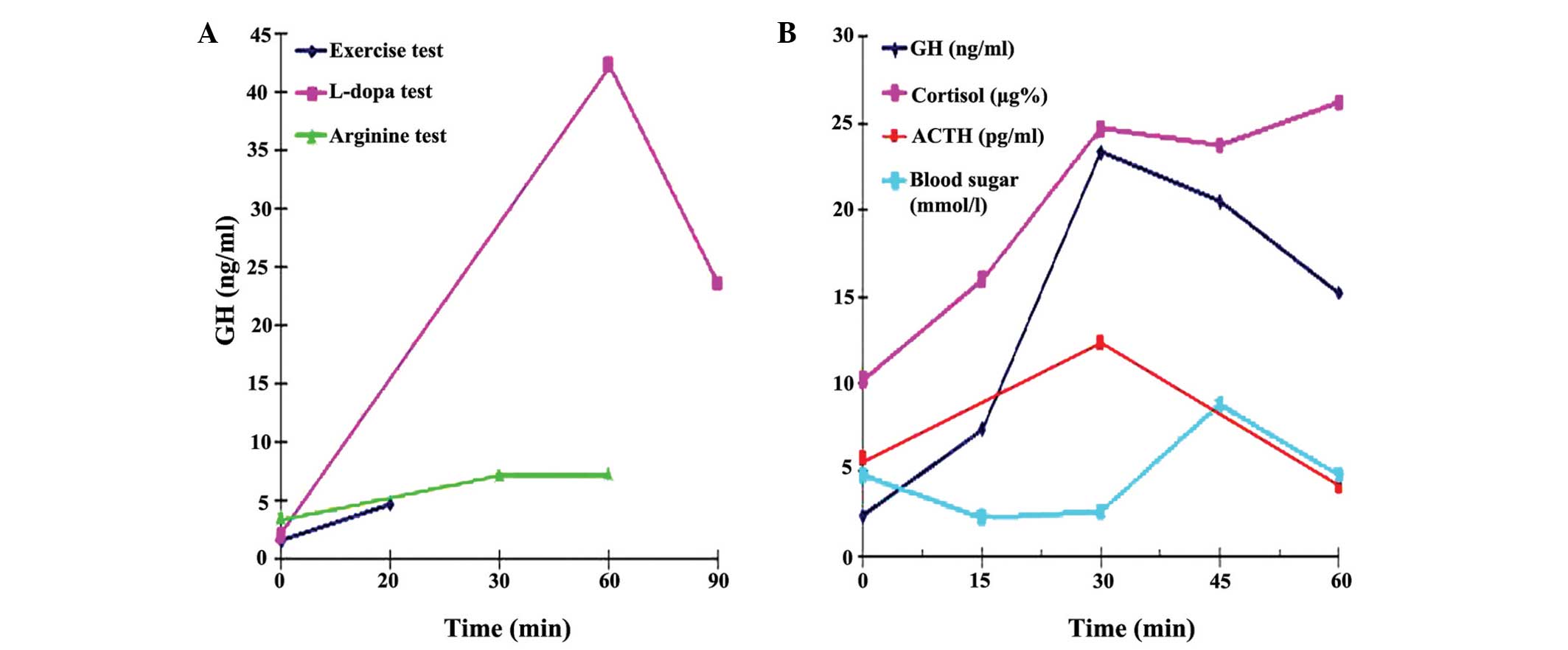
GH responses to the provocation tests are shown in
Fig. 2. The patient showed normal
responses to the GH provocative tests using L-dopa (peak GH
concentration, 42.38 ng/ml) and insulin (peak GH concentration was
increased to 23.29 ng/ml during hypoglycemia). However, a slightly
blunted response was observed for the GH provocation test with
arginine [peak GH concentration, 7.31 ng/ml (<10 ng/ml)], and
the GH level during exercise was low [4.8 ng/ml (<5 ng/ml)]. The
cortisol and ACTH responses to insulin-induced hypoglycemia were
normal.
Gene mutation analysis
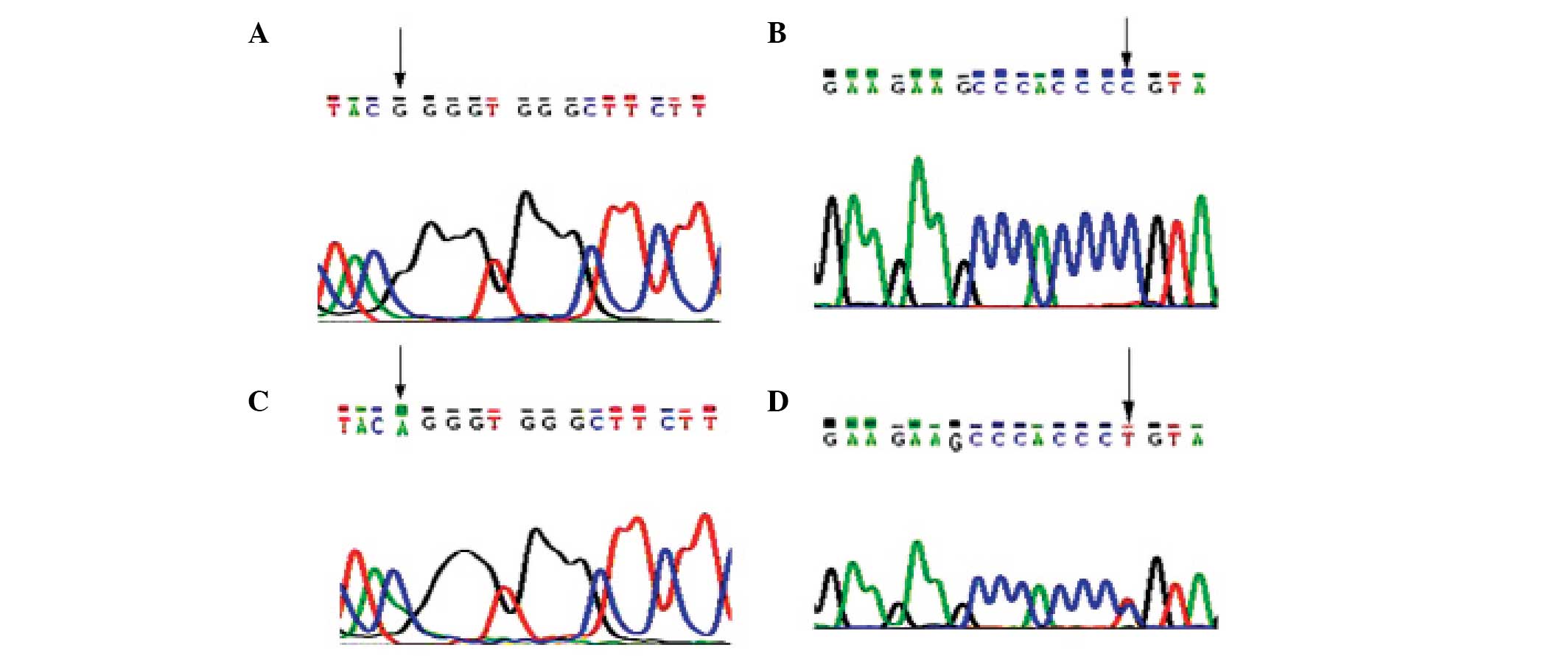
Gene mutation analysis revealed that the patient had
a G→A mutation at nucleotide 1,138 within exon 10 of the FGFR3
gene. This missense mutation caused the substitution of glycine
with arginine at amino acid position 380 (G380R) in the FGFR3
protein. The mutation was heterozygous, and the results of the
forward and reverse sequencing were consistent (Fig. 3). No mutation within exon 10 of the
FGFR3 gene was observed in the samples taken from the patient's
parents or from seven healthy controls (aged 10–35 years) from the
First Hospital of Lanzhou University.
Treatment and follow-up
A diagnosis of ACH with subnormal GH secretion was
considered on the basis of the clinical and laboratory results.
Oral therapy with recombinant human growth hormone (rhGH; 0.1
IU/kg/day) and levothyrocine (12.5 µg/day) was initiated. After six
months, the patient's height had markedly improved (to 93.5 cm).
Furthermore, the arm span-to-height ratio and lower limb
length-to-height ratio increased during treatment, while the head
circumference decreased. The serum GH concentration was 60.37
ng/ml. No abnormality was found on the thyroid function and sex
hormone tests.
Discussion
ACH, an autosomal dominant disorder, is the most
common form of human dwarfism. The main clinical manifestation is
an abnormally large head with a prominent forehead and flat nasal
bridge, short upper arms and legs (rhizomelic dwarfism), an
unusually prominent abdomen and buttocks, and short hands with
fingers that assume a trident or three-pronged position during
extension. In 1994, Francomano et al located the pathogenic
gene of ACH to chromosome 4p16.3 through genetic linkage analysis
(8). Soon after, Shiang et al
reported a missense mutation at codon 380 in the transmembrane
domain of the FGFR3 gene in patients with ACH (5,6). A G→A
displacement at nucleotide 1,138 in the FGFR3 gene is present in
90% of patients with ACH, while a G→C transversion is present in
only a minority of patients (9,10).
Similar results have been reported in Chinese populations (11). G375C and G346E mutations on the
transmembrane domain of the FGFR3 gene have also been observed in
patients with ACH (12,13). These results indicate a strong
association between the transmembrane domain of the FGFR3 gene and
ACH. Furthermore, Zhang et al reported a Ser217Cys mutation
in the Ig II domain of the FGFR3 gene in a Chinese family with ACH
(14).
FGFR3, a type of tyrosine receptor, comprises 806
amino acid residues and plays an important role in skeletal
development. The length of the FGFR3 gene is ~15 kb, with 19 exons
and 10 introns. Exon 10 encodes the transmembrane domain of the
FGFR3 gene. As a membrane receptor, the structure of FGFR3 is
comprised of three parts, including an intracellular region, a
transmembrane domain and an extracellular region, the latter of
which functions as a binding domain for numerous ligands, including
three typical Ig-like structural domains (Ig I–III) (15). The intracellular region includes a
near-membrane area, two conservative tyrosine kinase functional
domains and an autophosphorylated C-terminal. The ligand,
fibroblast growth factor (FGF), may attach to acetylated proteins
on the surface of the cells and induce receptor dimerization and
transautophosphorylation of the tyrosine kinase in the cytoplasm
(16). The residual phosphate
cellulose may be used as a docking site for proteins, subsequently
resulting in the activation of various signaling pathways that
mediate the effects of the FGF receptor with regard to the
regulation of proliferation, differentiation and apoptosis in a
number of cells (16).
In the present case report, the patient demonstrated
clinical manifestations similar to those described in the
literature on ACH, including a large head, prominent forehead,
short upper arms and legs, and short hands with fingers that
assumed a trident position. The results of the imaging examinations
were also consistent with a diagnosis of ACH. Finally, a G→A
displacement at nucleotide 1,138 within exon 10 of the FGFR3 gene
was identified, which further confirmed that nucleotide 1,138
within the FGFR3 gene is a common site of mutations leading to ACH.
The main reason that the FGFR3 mutation leads to ACH may be the
suppression of the proliferation and differentiation of cartilage
cells. The FGFR3 mutation causes dimerization of cell membrane
proteins, and may lead to the continuous activation of
intracellular tyrosine kinases in the absence of a ligand, which
ultimately activates intracellular signaling pathways and
suppresses the proliferation and differentiation of cartilage cells
(17,18). Matsushita et al reported that
the extracellular signal-regulated kinase/mitogen-activated protein
kinase signaling pathway in cartilage cells plays an important role
in the development of ACH (19).
Patients with ACH do not generally have a GH
deficiency (20). In the current
study, the patient demonstrated a normal response to GH provocation
tests with L-dopa and insulin; however, a blunted response was
observed in the GH provocation test with arginine (peak GH
concentration, <10 ng/ml). In addition, low GH concentrations
were observed during exercise (<5 ng/ml). Therefore, a subnormal
GH secretion was suspected. Furthermore, an MRI scan revealed that
the pituitary volume was decreased. Therefore, therapy with rhGH
(0.1 IU/kg/day) was initiated, and marked results were observed
within six months, which is consistent with previous studies
(21–24). A large-scale study in Japan
demonstrated that rhGH therapy promoted bone growth in patients
with ACH by improving the Z scores for growth rate and height, and
also had a dose- and time-dependent effect on ACH (25). A possible mechanism underlying the
effects of GH treatment on ACH may be through the epiphyseal growth
plate, which is the area of bone formation. GH stimulates local
cartilage cells in this area to produce insulin-like growth
factors, which promotes cartilage cell proliferation and thereby
promotes growth (26).
In the present study, no mutation was observed in
the FGFR3 gene of the patient's parents, indicating that the
patient had a de novo mutation. ACH has been associated with
advanced paternal age (2). As sperm
cells are produced constantly throughout life, the risk of
mutations in the sperm cells increases with age (21). This suggests that factors influencing
DNA replication and repair during spermatogenesis may predispose to
the occurrence of ACH-associated mutations. Therefore, the mutation
in the current patient was likely attributable to mutations in the
father's sperm cells.
In conclusion, the results of the present study
further support the hypothesis that the G1138A mutation within the
FGFR3 gene is the most common mutation causing ACH in various
populations. For certain ACH patients with subnormal GH secretion,
GH therapy may be beneficial for the treatment of short
stature.
References
|
1
|
Placone J and Hristova K: Direct
assessment of the effect of the Gly380Arg achondroplasia mutation
on FGFR3 dimerization using quantitative imaging FRET. PLoS One.
7:e466782012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orioli IM, Castilla EE and Barbosa-Neto
JG: The birth prevalence rates for the skeletal dysplasias. J Med
Genet. 23:328–332. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Horton WA, Hall JG and Hecht JT:
Achondroplasia. Lancet. 370:162–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Su N, Xu X, Li C, et al: Generation of
Fgfr3 conditional knockout mice. Int J Biol Sci. 6:327–332. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shiang R, Thompson LM, Zhu YZ, et al:
Mutations in the transmembrane domain of FGFR3 cause the most
common genetic form of dwarfism, achondroplasia. Cell. 78:335–342.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rousseau F, Bonaventure J, Legeai-Mallet
L, et al: Mutations in the gene encoding fibroblast growth factor
receptor-3 in achondroplasia. Nature. 371:252–254. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li YQ, Li QL and Zhang MH: Comparison of
diagnostic value of growth hormone exercise test and growth hormone
provocative test on growth hormone deficiency. J Appl Clin Pediatr.
24:601–602. 2009.
|
|
8
|
Francomano CA, Ortiz de Luna RI, Hefferon
TW, et al: Localization of the achondroplasia gene to the distal 2.
5Mb of human chromosome 4p. Hum Mol Genet. 3:787–792. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He X, Xie F and Ren ZR: Rapid detection of
G1138A and G1138C mutations of the FGFR3 gene in patients with
achondroplasia using high-resolution melting analysis. Genet Test
Mol Biomarkers. 16:297–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pehlivan S, Ozkinay F, Okutman O, et al:
Achondroplasia in Turkey is defined by recurrent G380R mutation of
the FGFR3 gene. Turk J Pediatr. 45:99–101. 2003.PubMed/NCBI
|
|
11
|
Cui Y, Zhao H, Liu Z, Liu C, Luan J, Zhou
X and Han J: A systematic review of genetic skeletal disorders
reported in Chinese biomedical journals between 1978 and 2012.
Orphanet J Rare Dis. 7:552012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Superti-Furga A, Eich G, Bucher HU, Wisser
J, Giedion A, Gitzelmann R and Steinmann B: A glycine
375-to-cysteine substitution in the transmembrane domain of the
fibroblast growth factor receptor-3 in a new born with
achondroplasia. Eur J Pediatr. 154:215–219. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prinos P, Kilpatrick MW and Tsipouras P: A
novel G346E mutation in achondroplasia. Pediatr Res.
37:151A1994.
|
|
14
|
Zhang SR, Zhou XQ, Ren X, et al: Ser217Cys
mutation in the Ig II domain of FGFR3 in a Chinese family with
autosomal dominant achondroplasia. Chin Med J (Engl).
120:1017–1019. 2007.PubMed/NCBI
|
|
15
|
Coutts JC and Gallagher JT: Receptors for
fibroblast growth factors. Immunol Cell Biol. 73:584–589. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mohammadi M, Dikic I, Sorokin A, Burgess
WH, Jaye M and Schlessinger J: Identification of six novel
autophosphorylation sites on fibroblast growth factor receptor 1
and elucidation of their importance in receptor activation and
signal transduction. Mol Cell Biol. 16:977–989. 1996.PubMed/NCBI
|
|
17
|
Webster MK and Donoghue DJ: Constitutive
activation of fibroblast growth factor receptor 3 by the
transmembrane domain point mutation found in achondroplasia. EMBO
J. 15:520–527. 1996.PubMed/NCBI
|
|
18
|
Naski MC, Wang Q, Xu J and Ornitz DM:
Graded activation of fibroblast growth factor receptor 3 by
mutations causing achondroplasia and thanatophoric dysplasia. Nat
Genet. 13:233–237. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsushita T, Wilcox WR, Chan YY, et al:
The FGFR3 promotes synchondrosis closure and fusion of ossification
centers through the MAPK pathway. Hum Mol Genet. 18:227–240. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka N, Katsumata N, Horikawa R and
Tanaka T: The comparison of the effects of short-term growth
hormone treatment in patients with achondroplasia and with
hypochondroplasia. Endocr J. 50:69–75. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanaka H, Kubo T, Yamate T, Ono T, Kanzaki
S and Seino Y: Effect of growth hormone therapy in children with
achondroplasia: growth pattern, hypothalamic-pituitary function,
and genotype. Eur J Endocrinol. 138:275–280. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramaswami U, Rumsby G, Spoudeas HA,
Hindmarsh PC and Brook CG: Treatment of achondroplasia with growth
hormone: six years of experience. Pediatr Res. 46:435–439. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamate T, Kanzaki S, Tanaka H, et al:
Growth hormone (GH) treatment in achondroplasia. J Pediatr
Endocrinol. 6:45–52. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Escamilla RF, Hutchings JJ, Li CH and
Forsham P: Achondroplastic dwarfism Effects of treatment with human
growth hormone. Calif Med. 105:104–110. 1996.
|
|
25
|
Seino Y, Yamanaka Y, Shinohara M, et al:
Growth hormone therapy in achondroplasia. Horm Res. 53:53–56. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wilkin DJ, Szabo JK, Cameron R, et al:
Mutations in fibroblast growth factor receptor 3 in sporadic cases
of achondroplasia occur exclusively on the paternally derived
chromosome. Am J Hum Genet. 63:711–716. 1998. View Article : Google Scholar : PubMed/NCBI
|