Introduction
Thrombosis is a pathogenically complex condition
with multiple risk factors, of which infection and vascular
inflammation are crucial (1,2). Various cytokines have been demonstrated
to influence thrombogenesis, such as tumor necrosis factor (TNF)-α
and interleukin (IL)-6, which are primarily secreted by activated
monocytes and macrophages (3).
Inflammation-induced TNF-α and IL-6 subsequently stimulate vascular
endothelial cells to become prothrombotic and exhibit anticoagulant
properties to various extents. Prothrombotic vascular endothelial
cells then express chemokines and E-selectin, which facilitate
thromobogenesis by increasing the ability of platelets to adhere to
vascular thrombosis sites via interaction with CX3CL1 or E-selectin
(4,5), or the induction of platelet aggregation
involving chemokine (C-C motif) ligand 5 (6,7).
Furthermore, cytokines are known to stimulate the secretion of
tissue factor from monocytes, macrophages and endothelial cells
(via TNF-α) (8,9), and to induce the expression of
plasminogen activator inhibitor-1 and C-reactive protein in the
liver (10–13). In addition, TNF-α and IL-6 have been
demonstrated to increase the risk of thrombosis development by
stimulating the secretion and inhibiting the cleavage of ultralarge
Von Willebrand multimers (14).
Upregulated proinflammatory cytokines in macrophages and
endothelial cells further promote the dysfunction of the
endothelium (15,16), forming a positive feedback signal.
Intravascular thrombosis is a well-recognized complication of
vascular inflammation. Thus, anti-inflammatory intervention in the
vasculature, particularly the vascular endothelium, may be an
effective strategy for the prevention or mitigation of
thrombosis.
Chebulagic acid (CA) is a key chemical component of
the traditional Mongolian anti-thrombotic drug Garidi-13 (17). The anti-infective and the
anti-inflammatory effects of CA have been recognized. CA has been
demonstrated to limit herpes simplex virus (HSV) infection by
targeting viral glycoproteins and inhibiting HSV-1 entry and
cell-to-cell transmission (18). HSV
infection has been confirmed to be involved in the pathogenesis of
atherosclerosis and thrombosis (19–22).
Furthermore, the anti-inflammatory effects of CA have been observed
to inhibit the activity of cyclooxygenase, a key thrombosis
promoter (23–26). In addition, CA has been demonstrated
to attenuate lipopolysaccharide (LPS)-induced inflammation by
suppressing nuclear factor (NF)-κB and mitogen-activated protein
kinase (MAPK) activation in macrophages (27). Therefore, the anti-inflammatory
effects of CA may be the basis of its anti-thrombotic potential,
and thus require further study.
In the present study, the ability of CA to inhibit
LPS-induced vascular inflammation was investigated by measuring the
levels of IL-1β and TNF-α in EA.hy926 human endothelial cells. The
underlying mechanism was also investigated.
Materials and methods
Reagents and cell cultures
Escherichia coli LPS and CA were purchased
from Sigma-Aldrich (St. Louis, MO, USA) and were resolved in
RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 2% fetal bovine serum (FBS). EA.hy926 human
endothelial cells were obtained from the American Type Culture
Collection (Rockville, MD, USA). EA.hy926 cells were propagated in
RPMI-1640 medium supplemented with 10% FBS (Gibco Life
Technologies, Rockville, MD, USA) at 37°C under 5% CO2,
or maintained in RPMI-1640 medium supplemented with 2% FBS.
EA.hy926 cells at ~85% confluence were treated with 0, 10, 100 or
1,000 ng/ml LPS and/or with 0, 10 or 50 µM CA for 0, 3, 6 or 12 h.
Cells were subsequently lysed for mRNA and protein expression
analysis. For the TNF-α and IL-1β assays, the supernatant of the
LPS- and/or CA-treated EA.hy926 cells was collected and centrifuged
at for 15 min at 4°C and 13,200 × g. The supernatant was then
transferred to new tubes and stored at −20°C until required for
assays.
ELISA assay for IL-1β or TNF-α
For the determination of the expression levels of
IL-1β and TNF-α, the supernatants of the LPS- and/or CA-treated
EA.hy926 cells were analyzed using an human IL-1β or TNF-α ELISA
kit (Shanghai ExCell Biology Inc., Shanghai, China) according to
the manufacturer's instructions. In brief, standards and samples
were diluted with phosphate-buffered saline (PBS), loaded onto a
96-well plate and incubated for 90 min at 37°C. Next,
biotin-labeled antibodies against IL-1β or TNF-α were utilized for
specific binding. Finally, an avidin-labeled enzyme and substrate
were used to quantitatively examine the levels of IL-1β and TNF-α
using a spectrophotometer (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Western blot analysis
EA.hy926 endothelial cells post treatment were
treated with ice-cold cell lysis reagent (Sigma-Aldrich) according
to the manufacturer's instructions, and each protein sample was
supplemented with protease inhibitor cocktail (Roche Diagnostics,
Basel, Switzerland), then quantified by bicinchoninic acid assay
(Pierce Biotechnology, Inc., Rockford, IL, USA). Protein samples
were separated using SDS-PAGE (10–12%) and detected by western blot
analysis using polyclonal rabbit antibodies against p38,
phosphorylated p38 (#sc-535, 1:500), c-Jun N-terminal kinase (JNK;
#sc-571, 1:400), phosphorylated JNK (#sc-135642, 1:200),
extracellular signal-regulated kinase (ERK), phosphorylated ERK
(#sc-23759-R, 1:200), TLR4 (#sc-10741, 1:200) and β-actin
(#sc-130656, 1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). Horseradish peroxidase-conjugated goat anti-rabbit IgG
secondary antibodies (Pierce) and an electrochemiluminescence
detection system (Amersham Pharmacia Biotech, Amersham, UK) were
used for detection.
mRNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total mRNA was extracted from cell samples using an
RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA) according to the
manufacturer's instructions, and supplemented with RNase inhibitor
(Takara Bio, Inc., Tokyo, Japan). RT-qPCR analysis of the TLR4 mRNA
level was performed using a QuantiTect SYBR Green PCR kit (Qiagen,
Inc.). All mRNA expression levels were normalized against β-actin.
The 2−∆∆Ct method was used for the relative
quantification of TLR4 mRNA expression (28).
Statistical analysis
Data are presented as the mean ± standard error, and
were analyzed using Student's t-test. P<0.05 was considered to
indicate a statistically significant difference. All tests were
performed using GraphPad Prism 6 software (GraphPad Software, Inc.,
La Jolla, CA, USA).
Results
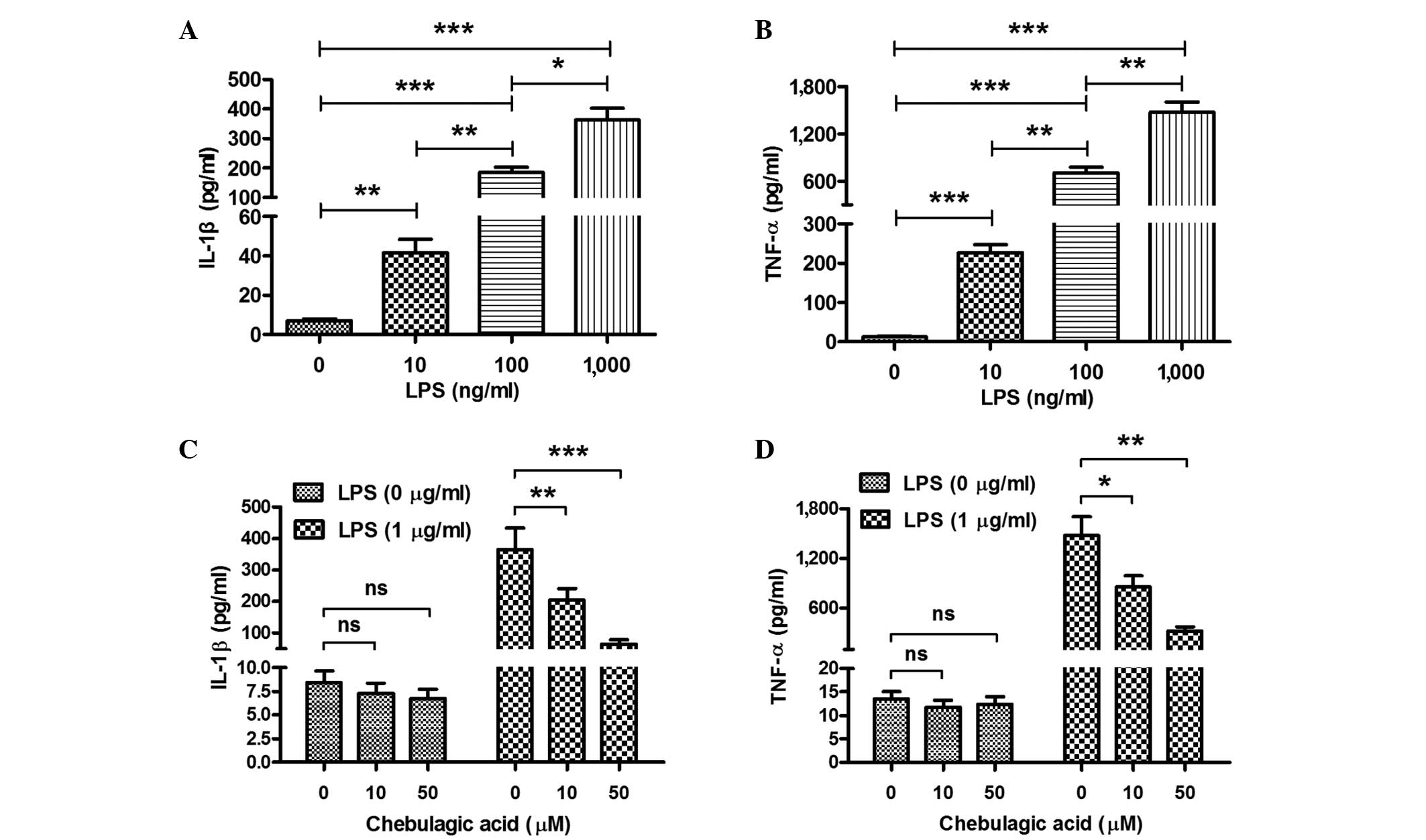
CA inhibits the LPS-induced secretion
of IL-1β and TNF-α in endothelial cells
In order to determine whether CA inhibits the
LPS-induced secretion of IL-1β and TNF-α, EA.hy926 endothelial
cells were treated with LPS (10, 100 or 1,000 ng/ml) and/or CA (0,
10 or 50 µM) for 12 h. Subsequently, the IL-1β and TNF-α content in
the cell supernatant was determined using an ELISA assay. Firstly,
it was demonstrated that the LPS treatment significantly promoted
the expression of IL-1β and TNF-α in the cell supernatant.
Treatment with 10 ng/ml LPS induced a 5.8-fold elevation in the
levels of IL-1β and a 19-fold elevation of TNF-α levels in the
EA.hy926 cells (P<0.01 and P<0.001, respectively; Fig. 1A and B). The promotion of IL-1β and
TNF-α secretion was dose-dependent; significant difference in the
levels of IL-1β and TNF-α were observed between the 10 and 100 or
1,000 ng/ml groups, as follows: 10 vs. 100 ng/ml (P<0.01) and
100 vs. 1,000 ng/ml (P<0.05) for IL-1β; 10 vs. 100 ng/ml and 100
vs. 1,000 ng/ml (P<0.01) for TNF-α. Secondly, the induction of
IL-1β and TNF-α in EA.hy926 cells was reexamined following combined
treatment with LPS and CA. CA was observed to significantly
attenuate the LPS-induced increase in the expression of IL-1β and
TNF-α (Fig. 1C and D). Reduced
levels of IL-1β or TNF-α were observed in EA.hy926 cells treated
with a combination of 1 µg/ml LPS and 10 or 50 µM CA compared with
the levels in EA.hy926 cells treated with 1 µg/ml LPS only
(P<0.01 and P<0.001, respectively, for IL-1β; P<0.05 and
P<0.01, respectively, for TNF-α). This attenuation was
dose-dependent, as the LPS-induced levels of IL-1β and TNF-α in the
cells treated with 50 µM CA were observed to be lower than those in
the cells treated with 10 µM CA. Notably, the inhibition of IL-1β
or TNF-α expression by CA was not significant in the EA.hy926 cells
that were not treated with LPS, possibly as the background
secretion of cytokines was insufficient to discriminate the
inhibitory effect of CA.
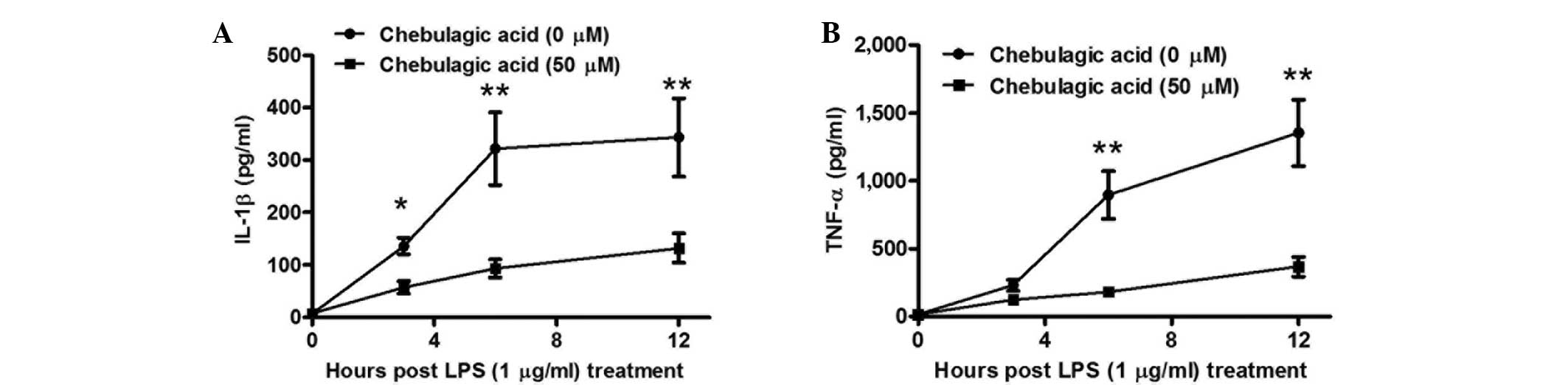
In order to further investigate the CA-mediated
inhibition of proinflammatory cytokines in endothelial cells, the
levels of IL-1β and TNF-α were determined in the supernatants of
EA.hy926 cells at various periods following treatment with 1 µg/ml
LPS and/or with 50 µM CA. As presented in Fig. 2A, the inhibitory effect of CA on
IL-1β secretion was significant at 3 h post-treatment. The levels
of IL-1β were found to be reduced in cells treated with a
combination of 50 µM CA and 1 µg/ml LPS compared with those in the
cells treated with LPS alone (P<0.05), and the inhibitory effect
was greater at 6 or 12 h post-treatment (P<0.01). Furthermore,
the inhibitory effect of TNF-α secretion was initially observed at
6 h post-treatment (P<0.01), then reconfirmed at 12 h
post-treatment (P<0.01). Therefore, the results indicate that
the CA-mediated inhibition of proinflammatory cytokines in the
endothelial cells was time-dependent.
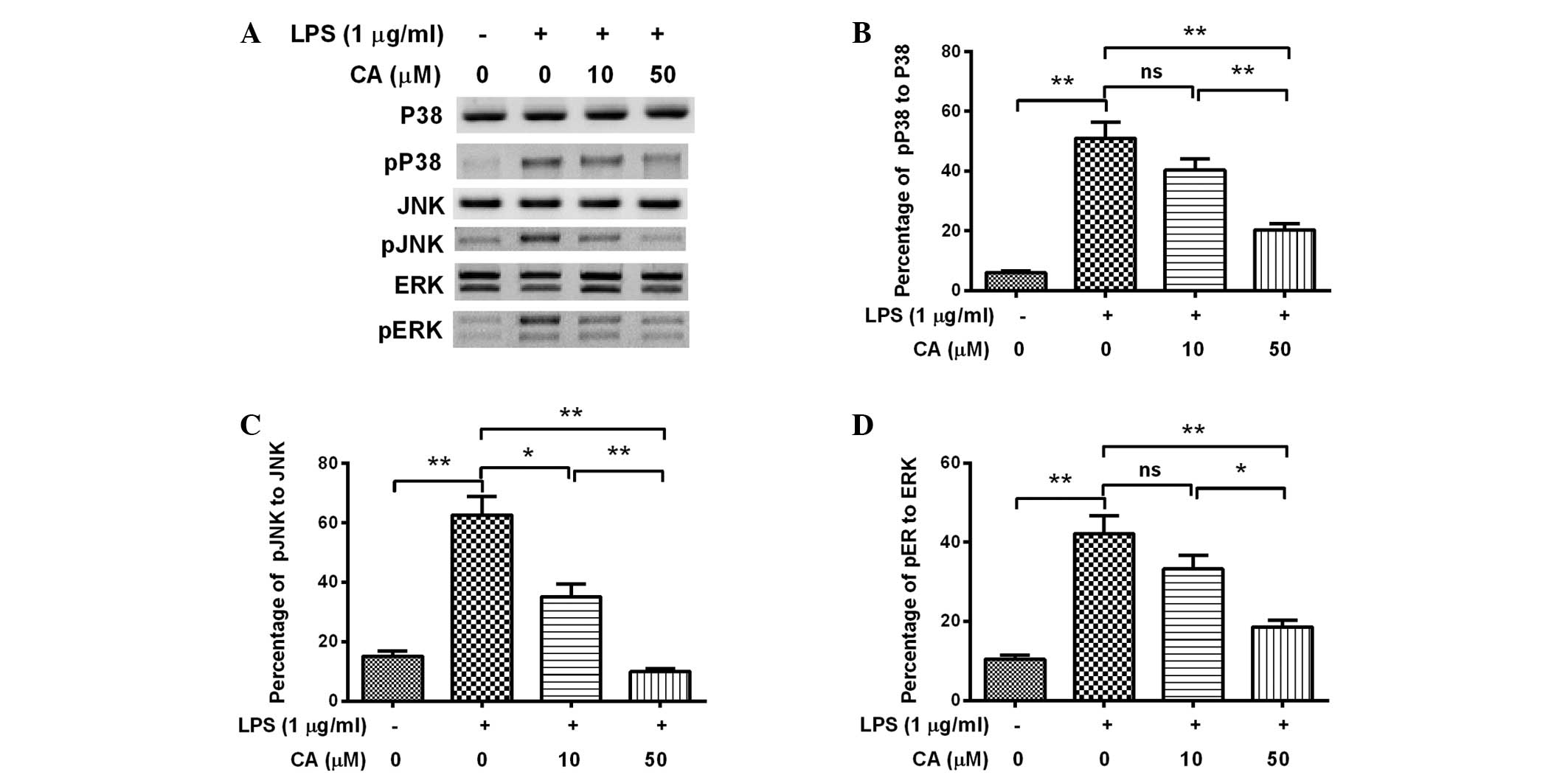
CA inhibits the LPS-induced activation
of MAPK signaling in endothelial cells
In order to investigate how CA affected the
secretion or expression of IL-1β or TNF-α, the degree of activation
of MAPK signaling in the LPS- and/or CA-treated EA.hy926 cells was
analyzed, as MAPK signaling is known to be promoted by LPS and to
upregulate proinflammatory cytokines (29,30).
Western blot analysis was performed to examine the expression and
phosphorylation of p38, JNK and ERK in EA.hy926 cells following
various treatments. Fig. 3 indicates
that levels of phosphorylated p38, JNK and ERK were significantly
elevated as a result of treatment with 1 µg/ml LPS for 12 h
(P<0.01). However, this promoted phosphorylation of p38, JNK and
ERK was significantly attenuated by treatment with 50 µM CA
(P<0.01), with JNK phosphorylation detectably attenuated by 10
µM CA (P<0.05). Furthermore, the attenuation was dose-dependent,
as a significant difference was detected between the 10 µM- and 50
µM-treated cells (P<0.01 for p38 or JNK phosphorylation;
P<0.05 for ERK phosphorylation). Therefore, the results indicate
that CA treatment attenuated MAPK activation in the LPS-induced
inflammatory cells.
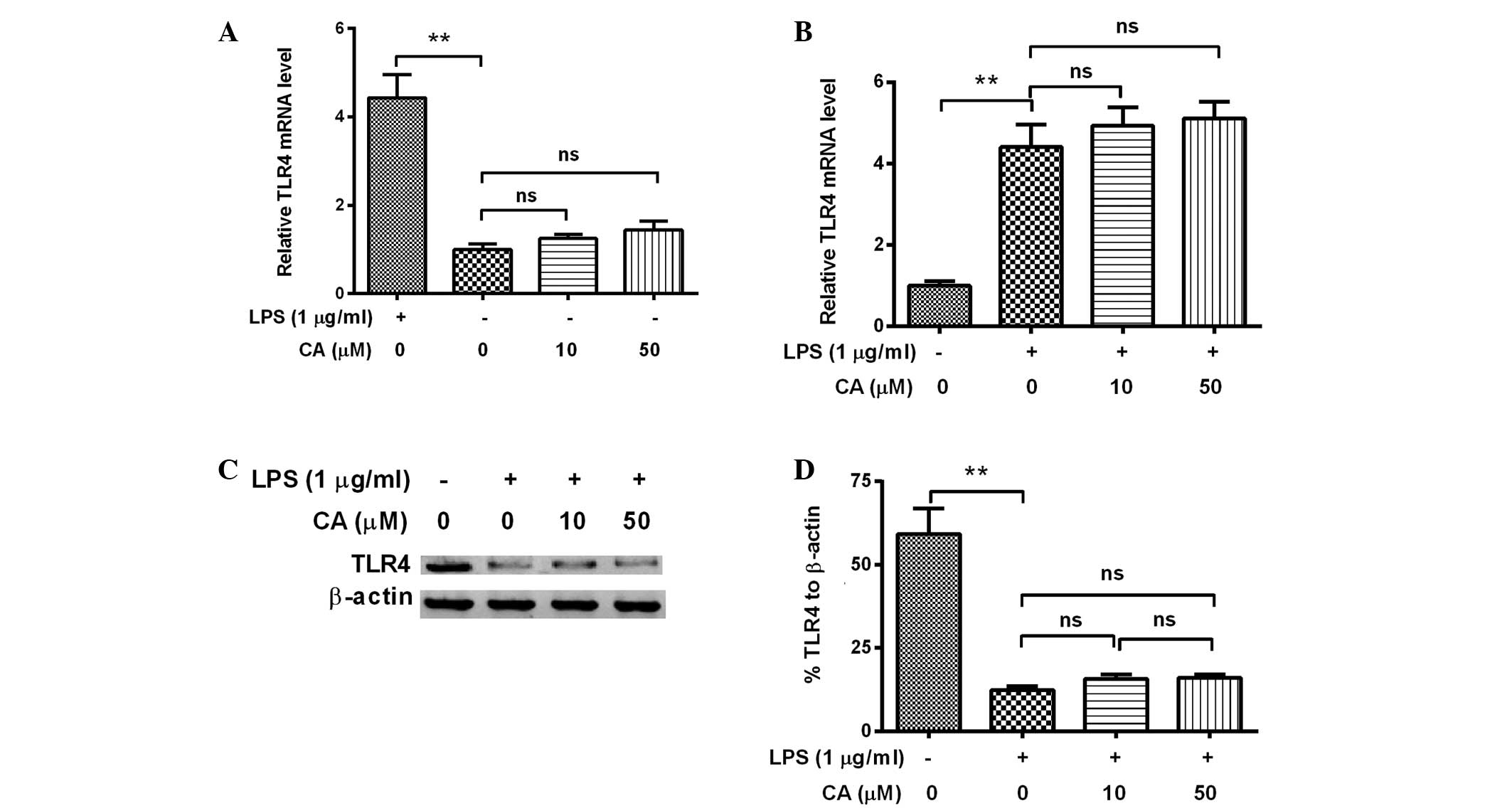
CA exerts no effect on the LPS-induced
activation of TLR4 signaling in endothelial cells
TLRs, such as TLR4, are type-I transmembrane
receptors expressed on the cell membrane following LPS stimulation
(31). Activation of TLR4 signaling
by LPS is associated with the release of LPS-induced inflammatory
cytokines (32). In order to
determine whether TLR4 signaling is targeted by CA to attenuate the
LPS-induced release of inflammatory cytokines, the expression of
TLR4 mRNA and protein was determined in EA.hy926 cells treated with
LPS and/or CA. As shown in Fig. 4A,
the mRNA expression levels of TLR4 were significantly upregulated
by LPS treatment (P=0.003); however, treatment with 10 or 50 µM CA
alone exerted no upregulatory effect on TLR4 mRNA (P=0.177 and
0.140, respectively). Furthermore, experiments in which combined
treatments were applied indicated that treatment with 10 or 50 µM
CA did not exert a significant effect on the LPS-induced promotion
of TLR4 mRNA levels (P=0.511 or 0.366; Fig. 4B). Western blot analysis indicated
that no CA-mediated regulation of TLR4 expression occurred at the
protein level in EA.hy926 cells, as no significant difference in
TLR4 expression levels was observed between the cells treated with
LPS alone and those treated with a combination of LPS and CA
(Fig. 4C and D).
Discussion
Endothelial inflammation has been implicated in a
variety of diseases, including infection (33), diabetes (34), atherosclerosis (35) and hypertension (36). Furthermore, endothelial inflammation
is commonly associated with thrombosis (37,38).
Various factors promote endothelial cells to an
inflammation-activated status and induce them to secrete
proinflammatory cytokines. Oncogenes and oxidative stress induce
vascular endothelial cells to exhibit distinct expression patterns
of proinflammatory cytokines (39).
In newly diagnosed type 2 diabetes, levels of serum IL-12 correlate
with endothelial dysfunction, insulin resistance and the expression
of proinflammatory cytokines (40).
Exposure to Shiga toxin 1, Shiga toxin 2, and α-sarcin induces
molecular damage and upregulates proinflammatory cytokines in human
endothelial cells (41).
Furthermore, classical swine fever virus has been found to induce
proinflammatory cytokine and tissue factor expression during the
establishment of long-term infection in porcine vascular
endothelial cells (42). In
addition, bacterial LPS modulates inflammasome gene expression and
regulates IL-1β and TNF-α secretion in endothelial cells (43,44).
Considering the high level of association of proinflammatory
cytokines, such as IL-1β and TNF-α, with thrombosis,
anti-inflammatory activity in the vasculature may be a response
strategy for the prevention or control of thrombosis.
The present study demonstrated the anti-inflammatory
effects of CA in endothelial cells. CA was demonstrated to inhibit
the LPS-induced secretion of two key proinflammatory cytokines,
IL-1β and TNF-α, in EA.hy926 endothelial cells. The LPS-induced
promotion of IL-1β and TNF-α in the EA.hy926 cells was
significantly attenuated by CA in a dose- and time-dependent
manner. Furthermore, it was observed that the CA inhibited
LPS-induced activation of MAPK signaling (29,30) in
endothelial cells. Western blot analysis demonstrated that the LPS
treatment promoted the phosphorylation of p38, JNK and ERK in
EA.hy926 cells. This promotion was inhibited by CA treatment, with
the phosphorylation of p38, JNK and ERK significantly attenuated by
treatment with ≥10 µM CA, in a dose-dependent manner.
TLRs, such as TLR4, are type-I transmembrane
receptors expressed on the cell membrane following LPS stimulation
(31). Activation of TLR4 signaling
by LPS is associated with the release of inflammatory cytokines
(32). However, no regulatory effect
of CA on TLR4 expression, which was upregulated by LPS during its
promotion of inflammatory cytokine release in EA.hy926 endothelial
cells, was observed. The RT-qPCR and western blot analyses
respectively revealed the upregulated mRNA and protein levels of
TLR4 by LPS in EA.hy926 cells were not significantly attenuated by
treatment with 10–50 µM CA. Therefore, it is hypothesized that the
regulation of IL-1β and TNF-α is targeted post TLR4 signaling.
However, the specific targets for CA remain unclear and require
identification.
In summary, the present study demonstrated that CA
inhibited LPS-induced vascular inflammation in EA.hy926 human
endothelial cells via the suppression of MAPK activation.
Acknowledgements
This study was supported by a grant from the Inner
Mongolia Science Foundation (no. 2012ms1211).
References
|
1
|
Richardson MW, Allen GA and Monahan PE:
Thrombosis in children: Current perspective and distinct
challenges. Thromb Haemost. 88:900–911. 2002.PubMed/NCBI
|
|
2
|
Gurgey A and Aslan D: Outcome of
noncatheter-related thrombosis in children: Influence of underlying
or coexisting factors. J Pediatr Hematol Oncol. 23:159–164. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Becarevic M, Ignjatovic S and Majkic-Singh
N: Deterioration of thromboses in primary antiphospholipid
syndrome: TNF-alpha and anti-annexin A5 antibodies. Clin Lab.
58:1079–1084. 2012.PubMed/NCBI
|
|
4
|
Schafer A, Schulz C, Eigenthaler M, et al:
Novel role of the membrane-bound chemokine fractalkine in platelet
activation and adhesion. Blood. 103:407–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raab M, Daxecker H, Markovic S, Karimi A,
Griesmacher A and Mueller MM: Variation of adhesion molecule
expression on human umbilical vein endothelial cells upon multiple
cytokine application. Clin Chim Acta. 321:11–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clemetson KJ, Clemetson JM, Proudfoot AE,
Power CA, Baggiolini M and Wells TN: Functional expression of CCR1,
CCR3, CCR4 and CXCR4 chemokine receptors on human platelets. Blood.
96:4046–4054. 2000.PubMed/NCBI
|
|
7
|
Bevilacqua MP, Pober JS, Majeau GR, Cotran
RS and Gimbrone MJ: Interleukin 1 (IL-1) induces biosynthesis and
cell surface expression of procoagulant activity in human vascular
endothelial cells. J Exp Med. 160:618–623. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Conway EM, Bach R, Rosenberg RD and
Konigsberg WH: Tumor necrosis factor enhances expression of tissue
factor mRNA in endothelial cells. Thromb Res. 53:231–241. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parry GC and Mackman N: Transcriptional
regulation of tissue factor expression in human endothelial cells.
Arterioscler Thromb Vasc Biol. 15:612–621. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Joseph L, Fink LM and Hauer-Jensen M:
Cytokines in coagulation and thrombosis: A preclinical and clinical
review. Blood Coagul Fibrinolysis. 13:105–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mantovani A, Sozzani S, Vecchi A, Introna
M and Allavena P: Cytokine activation of endothelial cells: New
molecules for an old paradigm. Thromb Haemost. 78:406–414.
1997.PubMed/NCBI
|
|
12
|
Esmon CT: Inflammation and thrombosis. J
Thromb Haemost. 1:1343–1348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tapper H and Herwald H: Modulation of
hemostatic mechanisms in bacterial infectious diseases. Blood.
96:2329–2337. 2000.PubMed/NCBI
|
|
14
|
Bernardo A, Ball C, Nolasco L, Moake JF
and Dong JF: Effects of inflammatory cytokines on the release and
cleavage of the endothelial cell-derived ultralarge von Willebrand
factor multimers under flow. Blood. 104:100–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smadja D, Gaussem P, Roncal C, Fischer AM,
Emmerich J and Darnige L: Arterial and venous thrombosis is
associated with different angiogenic cytokine patterns in patients
with antiphospholipid syndrome. Lupus. 19:837–843. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kowalska MA, Rauova L and Poncz M: Role of
the platelet chemokine platelet factor 4 (PF4) in hemostasis and
thrombosis. Thromb Res. 125:292–296. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han Q, Song J, Qiao C, Wong L and Xu H:
Preparative isolation of hydrolysable tannins chebulagic acid and
chebulinic acid from Terminalia chebula by high-speed
counter-current chromatography. J Sep Sci. 29:1653–1657. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin LT, Chen TY, Chung CY, et al:
Hydrolyzable tannins (chebulagic acid and punicalagin) target viral
glycoprotein-glycosaminoglycan interactions to inhibit herpes
simplex virus 1 entry and cell-to-cell spread. J Virol.
85:4386–4398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gyorkey F, Melnick JL, Guinn GA, Gyorkey P
and DeBakey ME: Herpesviridae in the endothelial and smooth muscle
cells of the proximal aorta in arteriosclerotic patients. Exp Mol
Pathol. 40:328–339. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Melnick JL, Petrie BL, Dreesman GR, Burek
J, McCollum CH and DeBakey ME: Cytomegalovirus antigen within human
arterial smooth muscle cells. Lancet. 2:644–647. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hajjar DP, Pomerantz KB, Falcone DJ,
Weksler BB and Grant AJ: Herpes simplex virus infection in human
arterial cells. Implications in arteriosclerosis. J Clin Invest.
80:1317–1321. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Visser MR, Tracy PB, Vercellotti GM,
Goodman JL, White JG and Jacob HS: Enhanced thrombin generation and
platelet binding on herpes simplex virus-infected endothelium. Proc
Natl Acad Sci USA. 85:8227–8230. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu Y, Ricciotti E, Scalia R, et al:
Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci
Transl Med. 4:132r–154r. 2012. View Article : Google Scholar
|
|
24
|
Armstrong PC, Kirkby NS, Zain ZN, Emerson
M, Mitchell JA and Warner TD: Thrombosis is reduced by inhibition
of COX-1, but unaffected by inhibition of COX-2, in an acute model
of platelet activation in the mouse. PLoS One. 6:e200622011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Gaetano G, Donati MB and Cerletti C:
Prevention of thrombosis and vascular inflammation: Benefits and
limitations of selective or combined COX-1, COX-2 and 5-LOX
inhibitors. Trends Pharmacol Sci. 24:245–252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Umar A, Boisseau M, Yusup A, Upur H,
Begaud B and Moore N: Interactions between aspirin and COX-2
inhibitors or NSAIDs in a rat thrombosis model. Fundam Clin
Pharmacol. 18:559–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reddy DB and Reddanna P: Chebulagic acid
(CA) attenuates LPS-induced inflammation by suppressing NF-kappaB
and MAPK activation in RAW 264.7 macrophages. Biochem Biophys Res
Commun. 381:112–117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kirchner S, Boldt S, Kolch W, et al: LPS
resistance in monocytic cells caused by reverse signaling through
transmembrane TNF (mTNF) is mediated by the MAPK/ERK pathway. J
Leukoc Biol. 75:324–331. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang JS, Kim HM, Choi IY, et al: DBM1285
suppresses tumor necrosis factor alpha production by blocking p38
mitogen-activated protein kinase/mitogen-activated protein
kinase-activated protein kinase 2 signaling pathway. J Pharmacol
Exp Ther. 334:657–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Medvedev AE, Lentschat A, Wahl LM,
Golenbock DT and Vogel SN: Dysregulation of LPS-induced Toll-like
receptor 4-MyD88 complex formation and IL-1 receptor-associated
kinase 1 activation in endotoxin-tolerant cells. J Immunol.
169:5209–5216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levy O: Innate immunity of the human
newborn: Distinct cytokine responses to LPS and other toll-like
receptor agonists. J Endotoxin Res. 11:113–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schouten M, Wiersinga WJ, Levi M and van
der Poll T: Inflammation, endothelium and coagulation in sepsis. J
Leukoc Biol. 83:536–545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hartge MM, Unger T and Kintscher U: The
endothelium and vascular inflammation in diabetes. Diab Vasc Dis
Res. 4:84–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rojas E, Rodriguez-Molina D, Bolli P, et
al: The role of adiponectin in endothelial dysfunction and
hypertension. Curr Hypertens Rep. 16:4632014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hirahashi J, Hishikawa K, Kaname S, et al:
Mac-1 (CD11b/CD18) links inflammation and thrombosis after
glomerular injury. Circulation. 120:1255–1265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Contreras JL, Eckstein C, Smyth CA, et al:
Activated protein C preserves functional islet mass after
intraportal transplantation: A novel link between endothelial cell
activation, thrombosis, inflammation and islet cell death.
Diabetes. 53:2804–2814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Suzuki E, Takahashi M, Oba S and
Nishimatsu H: Oncogene- and oxidative stress-induced cellular
senescence shows distinct expression patterns of proinflammatory
cytokines in vascular endothelial cells. Scientific World Journal.
2013:7547352013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mishra M, Kumar H, Bajpai S, Singh RK and
Tripathi K: Level of serum IL-12 and its correlation with
endothelial dysfunction, insulin resistance, proinflammatory
cytokines and lipid profile in newly diagnosed type 2 diabetes.
Diabetes Res Clin Pract. 94:255–261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brigotti M, Carnicelli D, Ravanelli E, et
al: Molecular damage and induction of proinflammatory cytokines in
human endothelial cells exposed to Shiga toxin 1, Shiga toxin 2 and
alpha-sarcin. Infect Immun. 75:2201–2207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bensaude E, Turner JL, Wakeley PR, et al:
Classical swine fever virus induces proinflammatory cytokines and
tissue factor expression and inhibits apoptosis and interferon
synthesis during the establishment of long-term infection of
porcine vascular endothelial cells. J Gen Virol. 85:1029–1037.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pontillo A, Girardelli M, Agostinis C,
Masat E, Bulla R and Crovella S: Bacterial LPS differently
modulates inflammasome gene expression and IL-1beta secretion in
trophoblast cells, decidual stromal cells and decidual endothelial
cells. Reprod Sci. 20:563–566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hu Y, Chen X, Duan H, Hu Y and Mu X:
Pulsatilla decoction and its active ingredients inhibit secretion
of NO, ET-1, TNF-alpha and IL-1 alpha in LPS-induced rat intestinal
microvascular endothelial cells. Cell Biochem Funct. 27:284–288.
2009. View
Article : Google Scholar : PubMed/NCBI
|