Introduction
Osteoarthritis (OA) refers to a group of
degenerative joint diseases, which may be initiated by joint
injuries, obesity, gene mutations and aging. Cartilage
extracellular matrix (ECM) is maintained by chondrocytes, which
synthesize aggrecan and collagen, providing mechanical strength and
flexibility to joints. Osteoarthritic chondrocytes exhibit an
altered metabolism and an imbalance between anabolic growth factors
and proinflammatory cytokines, including tumor necrosis factor
(TNF)-α and interleukin (IL)-1, which are produced by inflammatory
cells, synovial fibroblasts and chondrocytes. Chondrocytes are a
distinct cell type that are targets for proinflammatory cytokines
during the pathogenesis of OA. These cytokines induce the
expression of matrix metalloproteinases (MMPs), which subsequently
cleave various components of the ECM (1–4).
However, the mechanisms by which TNF-α increases the expression of
MMP-13 in chondrocytes are yet to be fully elucidated.
Previous studies have indicated that low-density
lipoprotein receptor-related protein 1 (LRP1) may regulate cell
survival (5) and inflammation
(6). LRP1 is hypothesized to
regulate lipid homeostasis, extracellular proteolysis, growth
factor activity, the composition of the ECM and immune responses
(7). A significant number of these
LRP1 functions are considered to be associated with endocytosis and
cellular signal transduction pathways (8,9). Overton
et al (10) proposed that the
activity of LRP1 in atherosclerosis may be associated with its
ability to suppress local inflammation. Furthermore, LRP1
suppresses the expression of inflammatory mediators indirectly via
the regulation of TNF receptor 1 (TNFR1)-dependent cell signaling
through the IκB kinase nuclear factor (IKK-NF)-κB pathway (11). Similarly, Gaultier et al
(12) reported that LRP1 may inhibit
inflammation in cases of peripheral nerve injury.
However, the mechanisms by which LRP1 regulates
inflammation and apoptosis in chondrocytes remain unclear. In the
present study, it was hypothesized that LRP1 protected chondrocytes
against apoptosis and TNF-α-induced inflammation. Therefore, the
aim of the present study was to define the role of LRP1 in
TNF-α-induced apoptosis and inflammation of articular chondrocytes
in vitro. Thus, the mechanisms may identify a novel target
for attenuating catabolic TNF-α activity in cartilage tissue.
Materials and methods
Antibodies and reagents
Rabbit monoclonal antibodies targeting LRP1
(#2703-1; 1:1,000) were obtained from Epitomics, Inc. (Burlingame,
CA, USA), while monoclonal rabbit antibodies against glyceraldehyde
3-phosphate dehydrogenase (GAPDH; #2118; 1:1,000), phosphorylated
NF-κB subunit p65/RelA (#3033; 1:1,000) and mouse monoclonal
antibodies against IκB (#4814; 1:1,000) and were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). In addition,
rabbit polyclonal antibodies targeting Akt (#BS2987; 1:700),
phospho (p)-Akt (#BS4009; 1:600), B-cell lymphoma 2 (Bcl-2;
#BS1511; 1:600), Bcl-2-associated X protein (Bax; #BS1030; 1:600)
and caspase-3 (#BS1518; 1:700) were purchased from Bioworld
Technology, Inc. (St. Louis Park, MN, USA), and rabbit polyclonal
antibodies against MMP-13 (#sc-30073; 1:600) and nitric oxide
synthase (iNOS; #sc-67003; 1:600) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Dulbecco's modified Eagle's
medium (DMEM)/F-12 nutrient mixture and fetal bovine serum (FBS)
were purchased from Gibco Life Technologies (Carlsbad, CA, USA).
Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and
anti-goat IgG secondary antibodies were purchased from Wuhan Boster
Biological Technology, Ltd. (Wuhan, China). Recombinant rat TNF-α
was purchased from PeproTech, Inc. (Rocky Hill, NJ, USA). An NF-κB
inhibitor (Bay 11–7082) was purchased from the Beyotime Institute
of Biotechnology (Haimen, China). This study was approved by the
Ethics Committee of Wuhan University (Wuhan, China).
Cell lines
Primary chondrocytes were isolated from the knee
joint of rats (age, 7 days) using a sequential proteinase and
collagenase digestion technique (13), and the cells released by enzymatic
digestion were cultured to a density of 3.0×106 cells/ml
in DMEM/Ham's F-12 media (1:1) supplemented with 10% FBS (complete
media). The cells were cultured at 37°C in humidified air with 5%
CO2. The cells were maintained at 20–80% confluency, and
cells between passages 2 and 3 were used for the following
experiments. Subsequently, the cells were plated onto a six-well
plate at a density of 2.0×105 cells/ml. The chondrocytes
were starved in DMEM overnight, after which pretreatment with the
Bay 11–7082 NF-κB inhibitor (10 µM) was conducted 30 min prior to
the addition of TNF-α. With regard to the signaling experiments,
chondrocytes were treated with TNF-α (30 ng/ml) for 30 min. For the
quantitative polymerase chain reaction (qPCR) experiments, the
cells were treated with TNF-α (30 ng/ml) for 12 h.
Construction of a lentiviral vector
expressing LRP1-targeting small hairpin (sh)RNA
Knockdown of LRP1 expression in the chondrocytes was
achieved using a lentivirus vector expressing a rat LRP1-targeting
shRNA (shLRP1; Genechem, Shanghai, China), according to the
manufacturer's instructions. The target nucleotide sequence of the
oligoduplexes was as follows: 5′-GCA UUG GCG UGC AGC UUA AUU-3′. A
non-targeting control (NTC) oligoduplex was used as a negative
control. At a multiplicity of infection of 20:1 for 12 h after
transfection, the medium was exchanged for fresh medium, and the
cells were cultured for a further 48–72 h. mRNA was harvested after
48 h and protein was harvested after 72 h. The effects of LRP1
knockdown on the expression of various indices were determined
using qPCR and western blot analysis.
Western blot analysis
Protein was extracted from stably transfected cells
using a radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology), according to the manufacturer's
instructions. The protein concentration was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology), after which 30 µg protein per lane was loaded onto
a 10% SDS-PAGE gel. Proteins were transferred to a polyvinylidene
difluoride membrane, and blocked with 5% non-fat dry milk in
Tris-buffered saline and Tween 20 (TBST). The membranes were
incubated with the primary antibodies overnight at 4°C, and
subsequently washed three times with TBST. Next, the membranes were
incubated with the HRP-conjugated secondary antibodies (1:5,000) in
TBST for 2 h at room temperature. After washing with TBST buffer,
the western blots were visualized using an enhanced
chemiluminescence system (Thermo Fisher Scientific, Waltham, MA,
USA), and exposed to Kodak X-ray film (Kodak, Rochester, NY,
USA).
Reverse transcription (RT)-qPCR
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA), according to the
manufacturer's instructions. First-strand cDNA synthesis was
conducted using an RT-PCR kit (Takara Biotechnology Co., Ltd.,
Dalian, China), according to the manufacturer's instructions.
Target gene mRNA expression levels were determined using a SYBR
Premix Ex Taq (Takara Biotechnology Co., Ltd.) qPCR system, with
the following protocol: Denaturation for 10 sec at 95°C, followed
by 40 cycles of 15 sec at 95°C and 60 sec at 60°C. Forward and
reverse primer sequences are presented in Table I. GAPDH from the same sample was used
as the internal control, and the relative contents of the copy
numbers of the target gene mRNA were calculated. The qPCR
experiments were conducted independently and in triplicate.
 | Table I.Oligonucleotide primers used for
quantitative polymerase chain reaction analysis. |
Table I.
Oligonucleotide primers used for
quantitative polymerase chain reaction analysis.
| Gene | Accession number | Primer sequence |
|---|
| GAPDH |
|
|
|
Forward | NC_005103.3 |
ACAGCAACAGGGTGGTGGAC |
|
Reverse |
|
TTTGAGGGTGCAGCGAACTT |
| LRP1 |
|
|
|
Forward | NM_001130490 |
GACTAACCCCTGTGACCGCAA |
|
Reverse |
|
GGTGCCATTGTCCAGCCTCTT |
| TNFR1 |
|
|
|
Forward | NC_005103.3 |
TCAAAGAGGTGGAGGGTGAAGGA |
|
Reverse |
|
AGACAGGATGACTGAAGCGTGGG |
| iNOS |
|
|
|
Forward | NC_005109.3 |
AAAATGGTTTCCCCCAGTTC |
|
Reverse |
|
GCTTGTCTCTGGGTCCTCTG |
Determination of the levels of MMP-13
and collagen type II α 1 (COL2A1) using ELISA
Control and shLRP1 chondrocytes were pretreated with
Bay 11–7082 (10 µM) for 2 h and incubated with or without TNF-α (30
ng/ml) for a further 24 h. The effect of TNF-α on the expression
levels of MMP-13 and COL2A1 secreted by the chondrocytes in the
culture medium was assessed using a rat MMP-13 and COL2A1 ELISA kit
(Wuhan Elabscience Biotechnology Co., Ltd., Wuhan, China),
according to the manufacturer's instructions.
TUNEL assay
Control and shLRP1 chondrocytes were incubated with
or without TNF-α (30 ng/ml) for 24 h. Apoptotic cells were detected
using a TUNEL assay (Roche Diagnostics, Basel, Switzerland),
according to the manufacturer's instructions. Briefly, the cells
were fixed in 4% paraformaldehyde solution at 4°C for 1 h and
washed three times with fresh phosphate-buffered saline (PBS). The
slides were incubated with 50 µl terminal deoxynucleotidyl
transferase and TUNEL Reaction mixture for 1 h at 37°C in the dark.
2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (Beyotime
Institute of Biotechnology) solution was added to stain cell
nucleus for 5 min. After washing with PBS for 5 times with 3 min
interval, slides were covered with anti-fade mounting medium. Three
fields were obtained from each group using a fluorescence
microscope (Nikon Corporation, Tokyo, Japan).
Statistical analysis
Statistical evaluations were conducted using SPSS
software (SPSS, Inc., Chicago, IL, USA). Data are presented as the
mean ± standard deviation. The statistical significance between the
differences in the mean values was assessed using one-way analysis
of variance, where P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of LRP1 knockdown on the
chondrocytes
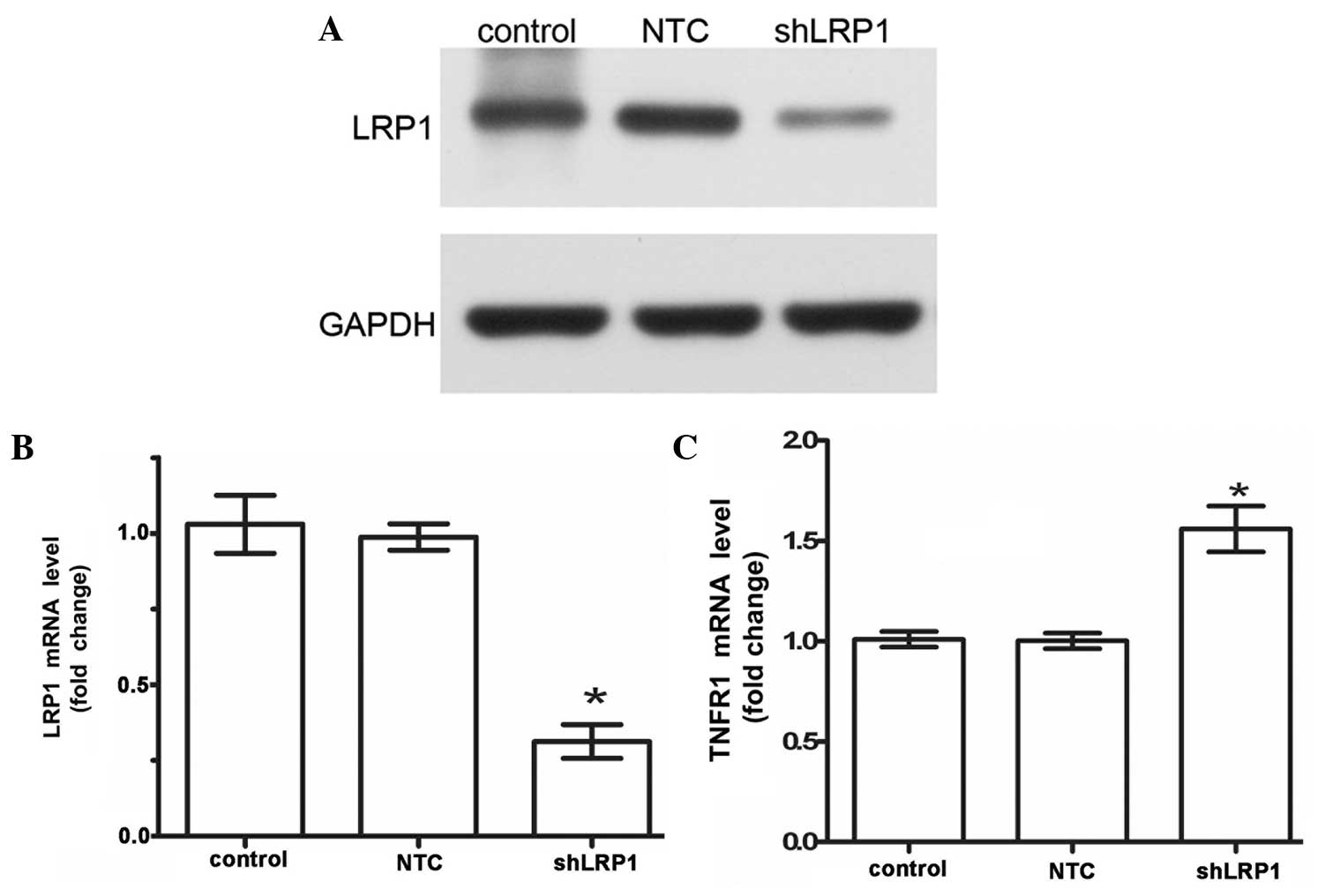
In order to knockdown LRP1 expression, RNA
interference technology was used to transfect the chondrocytes with
a lentivirus vector expressing an LRP1-targeting shRNA. The
transfection efficiency was ~80%, as determined by fluorescence
microscopy. Thus, the expression levels of LRP1 protein and mRNA
were substantially knocked down to 70% of the control (control,
1.020±0.01826; NTC, 0.9875±0.02175; shLRP1, 0.3125±0.0175; Fig. 1A and B). Furthermore, the
cell-surface mRNA expression levels of TNFR1 were measured using
qPCR, and the TNFR1 mRNA expression levels were determined to be
higher in the shLRP1 cells when compared with the control cells
(control, 1.010±0.01958; NTC, 1.003±0.01931; shLRP1, 1.560±0.057;
Fig. 1C). These results indicated
that the mRNA knockdown was specific for LRP1.
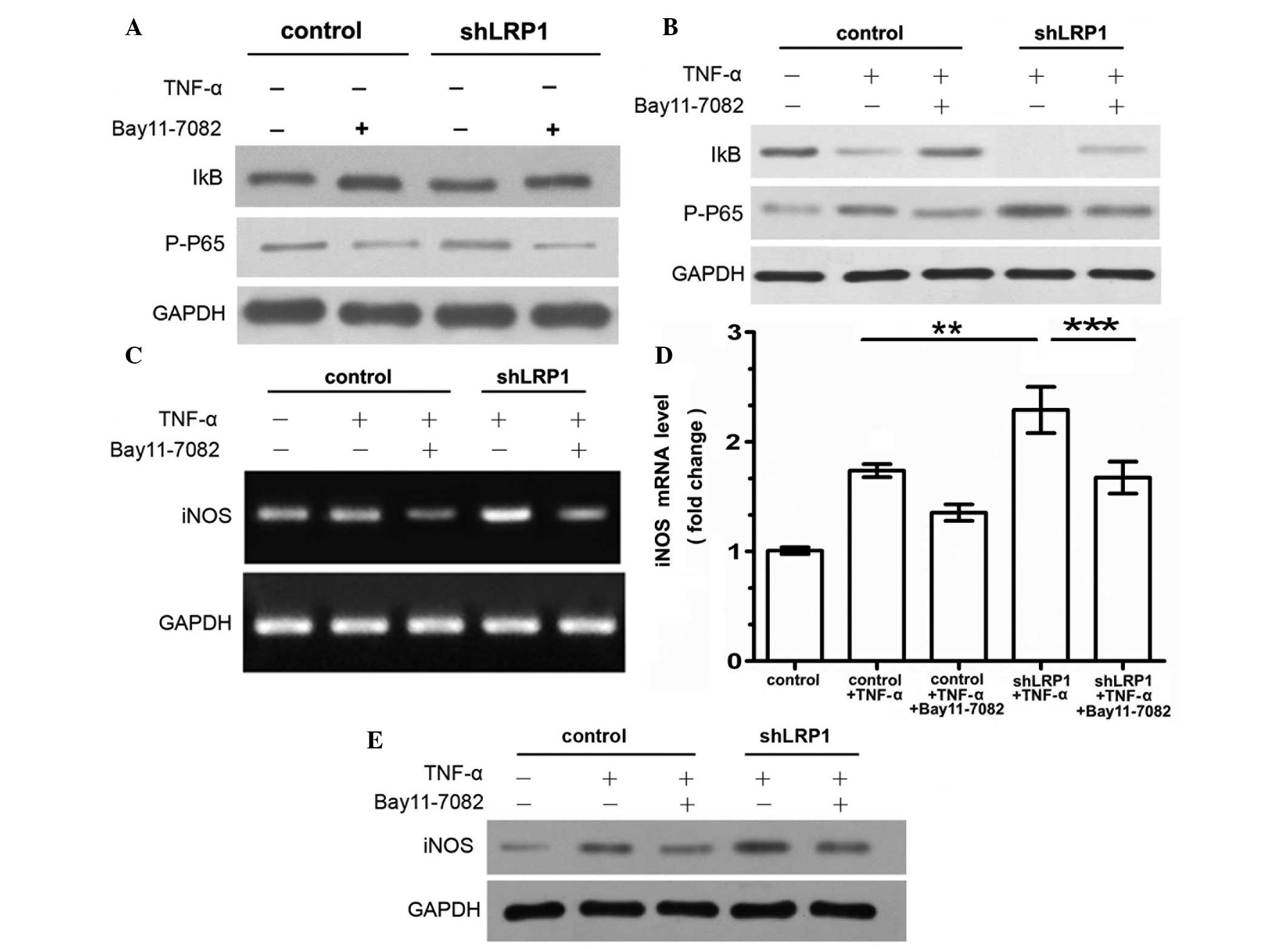
IKK-NF-κB pathway is activated by LRP1
knockdown in chondrocytes
NF-κB is a key mediator of TNF-α signal transduction
in chondrocytes (4). Therefore, the
effect of LRP1 on TNF-α-induced NF-κB activation and IκB
phosphorylation was evaluated in chondrocytes using western blot
analysis. IκB binds NF-κB, retaining it in the cytoplasm.
Phosphorylation of IκB promotes IκB the degradation and release of
activated NF-κB. In order to evaluate the role of the IKK-NF-κB
signaling pathway in mediating inflammation, the basal condition of
the IKK-NF-κB pathway was determined by treating the chondrocytes
with an NF-κB-specific inhibitor (Bay 11–7082) for 30 min, with no
exogenous stimulants. No statistically significant differences were
observed between the control and shLRP1 groups with regard to the
basal levels of the phosphorylated NF-κB subunit p65/RelA and IκB
protein (Fig. 2A). Next, the
activity of the IKK-NF-κB pathway was analyzed in chondrocytes
treated with Bay 11–7082 for 30 min prior to TNF-α stimulation.
Protein expression of phosphorylated p65/RelA was observed in the
control and LRP1-knockdown cells exposed to TNF-α, although
increased levels of the phosphorylated NF-κB subunit p65/RelA were
observed and IκB was undetectable in the shLRP1 cells. Furthermore,
Bay 11–7082 was demonstrated to inhibit the upregulation of
phosphorylated p65/RelA in the shLRP1 chondrocytes treated with
TNF-α (Fig. 2B).
Knockdown of LRP1 increases
TNF-α-induced expression of iNOS
Experiments were conducted to determine whether the
knockdown of LRP1 promoted the enhanced transcription of iNOS, as a
result of NF-κB activation. The results revealed that the mRNA
expression levels of iNOS were elevated in the shLRP1 chondrocytes
treated with TNF-α, as compared with the control cells.
Furthermore, the mRNA and protein expression levels of iNOS were
significantly reduced in the Bay 11–7082-pretreated shLRP1
chondrocytes exposed to TNF-α (shLRP1 + TNF-α, 2.29±0.12; control +
TNF-α, 1.737±0.03; shLRP1 + TNF-α + Bay 11–7082, 1.67±0.084;
Fig. 2C–E), providing evidence for
the activation of NF-κB in the LRP1 knockdown cells.
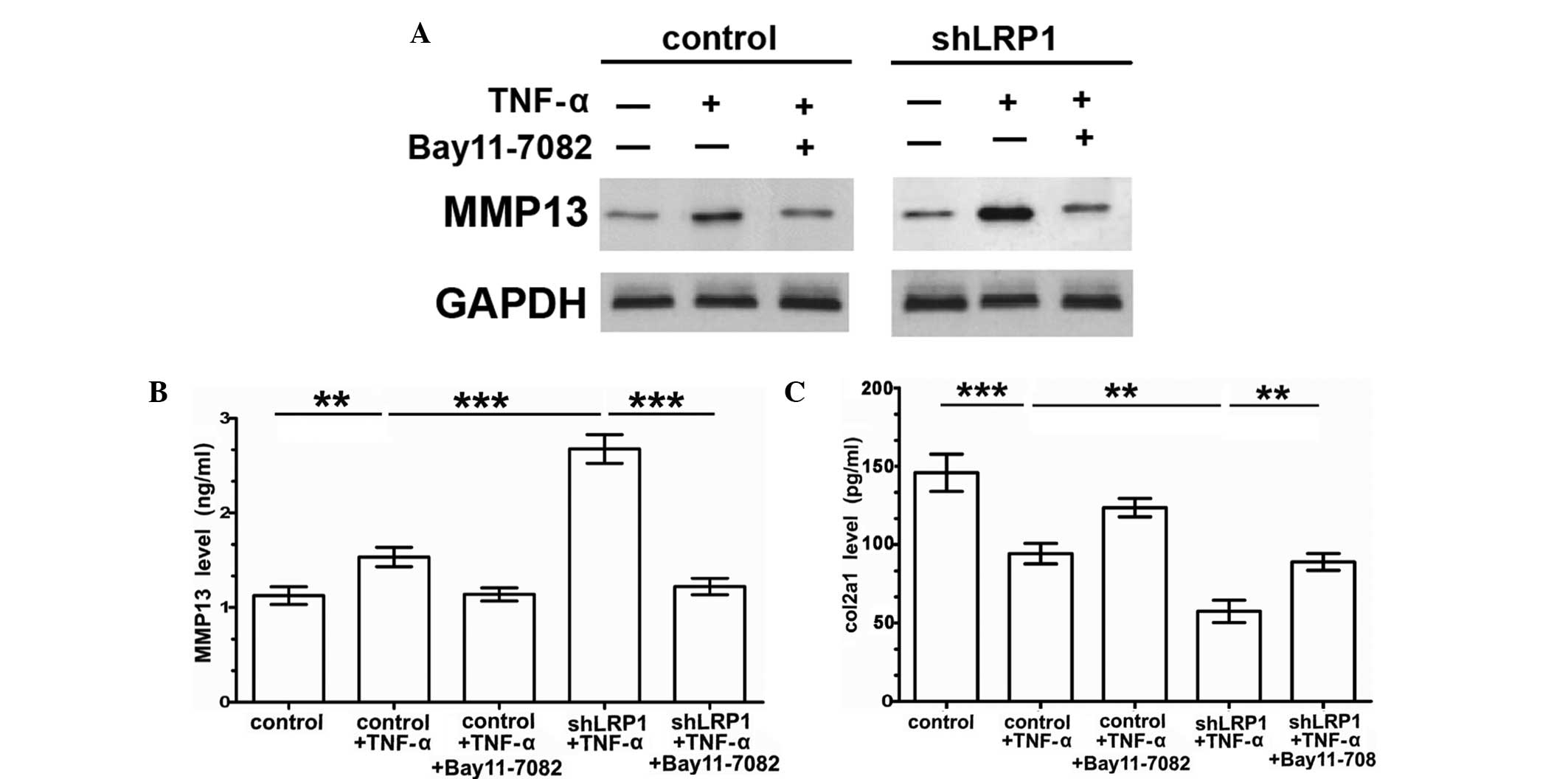
Knockdown of LRP1 increases
TNF-α-induced MMP-13 expression by activating the NF-κB
pathway
The IKK-NF-κB pathway is one of a number of key
TNF-α signal transducers in chondrocytes, and the signaling pathway
reportedly regulates the transcription of MMP-13 (4). Therefore, the effects of LRP1 on
TNF-α-induced MMP-13 expression were investigated. In the absence
of exogenous stimulants, no statistically significant alterations
were observed in the base protein expression levels of MMP-13
between the control and shLRP1 groups. However, MMP-13 expression
was substantially increased in the shLRP1 chondrocytes stimulated
with TNF-α (Fig. 3A). ELISA analysis
was used to determine the MMP-13 expression levels in the
chondrocyte culture medium. As shown in Fig. 3B, the extracellular levels of MMP-13
were increased in the TNF-α-induced shLRP1 chondrocyte medium.
Furthermore, the results indicated that the levels of MMP-13 were
significantly reduced in the Bay 11–7082-pretreated shLRP1 cells
that were exposed to TNF-α (control, 1.125±0.54; shLRP1 + TNF-α,
2.675±0.088; control + TNF-α, 1.533±0.059; shLRP1 + TNF-α + Bay
11–7082, 1.221±0.05). In addition, extracellular levels of COL2A1
were significantly downregulated in the TNF-α-induced shLRP1
chondrocytes (control, 145.8±6.85; shLRP1 + TNF-α, 57.44±4.12;
control + TNF-α, 94.26±3.754; shLRP1 + TNF-α + Bay 11–7082,
89.01±3.1; Fig. 3C), which was
reversed with Bay 11–7082 pretreatment. These results indicated
that LRP1 inhibited TNF-α-induced MMP-13 expression and indirectly
upregulated the extracellular levels of COL2A1 via the inhibition
of NF-κB.
Knockdown LRP1 chondrocytes are
sensitive to TNF-α-induced apoptosis due to the upregulation of Bax
and caspase-3 and the inhibition of p-Akt and Bcl-2
Since LRP1 was demonstrated to markedly affect
TNF-α-induced inflammation, the effects of LRP1 on apoptosis were
investigated by analyzing the levels of Akt, Bcl-2, Bax and
caspase-3 using western blot analysis and a TUNEL assay. Activation
of Akt and Bcl-2 has been previously associated with the inhibition
of the apoptotic cleavage of caspase-3 (14). To clarify this mechanism,
chondrocytes were cultured in serum free medium for 12 h and
treated with TNF-α or DMEM for an additional 2 h. The results
indicated that TNF-α-induced shLRP1 chondrocytes exhibited
significantly upregulated expression levels of caspase-3 (control,
0.1197±0.002; shLRP1 + TNF-α, 0.294±0.006; control + TNF-α,
0.117±0.003; shLRP1, 0.224±0.003) and reduced ratios of p-Akt/Akt
(control, 1.5±0.049; shLRP1 + TNF-α, 0.71±0.051; control + TNF-α,
1.326±0.061; shLRP1, 0.963±0.05; Fig.
4A) and Bcl-2/Bax (control, 3.145±0.05; shLRP1 + TNF-α,
1.309±0.048; control + TNF-α, 2.745±0.056; shLRP1, 1.574±0.041;
Fig. 4B). These results indicated
that LRP1 knockdown enhanced Bax and caspase-3 expression, while
reducing Bcl-2 expression, which may serve an antiapoptotic
function in chondrocytes.
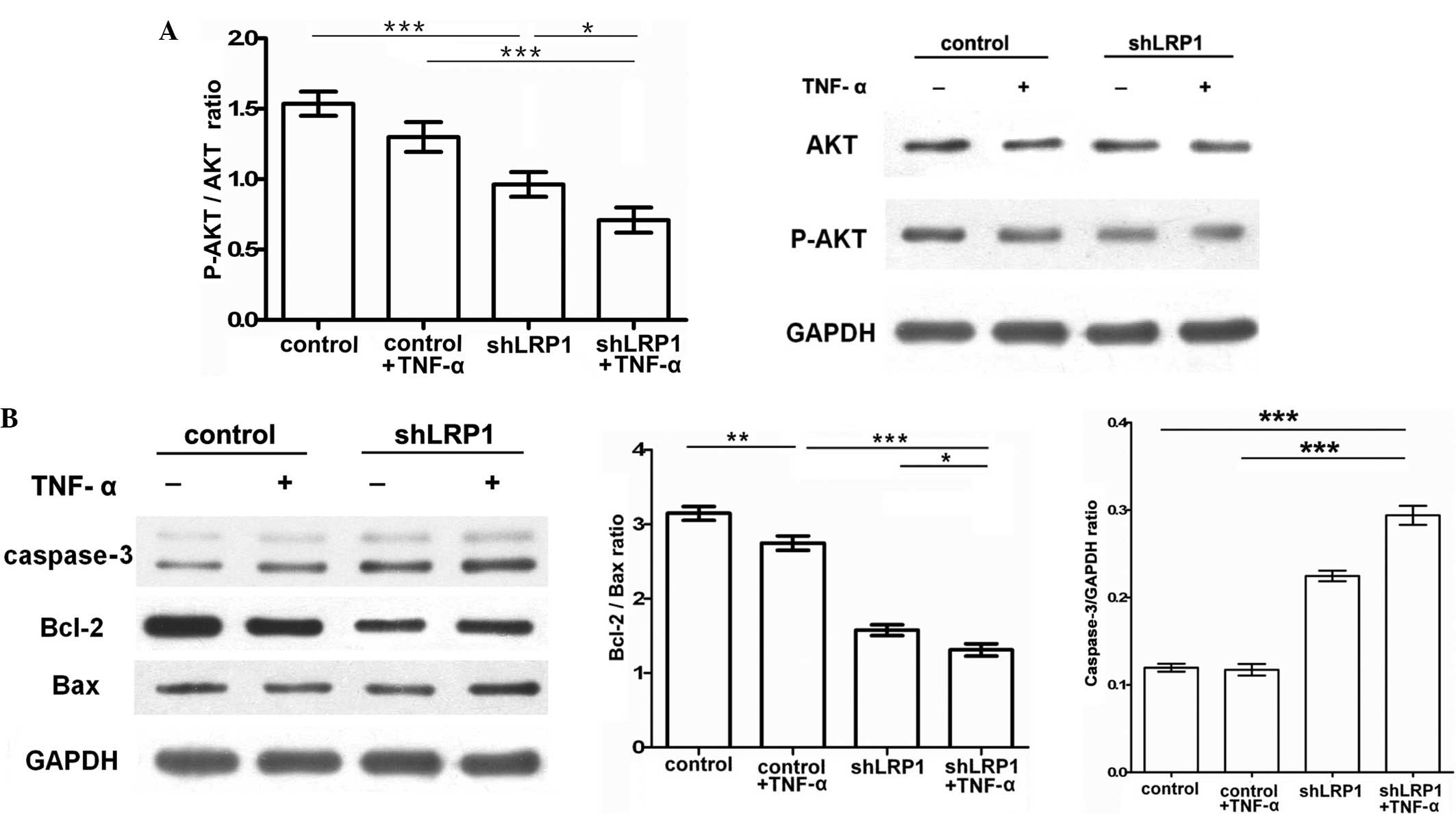 | Figure 4.LRP1 knockdown inhibits TNF-α-induced
activation of the phosphoinositide 3-kinase/Akt signaling pathway
in chondrocytes. Following starvation in Dulbecco's modified
Eagle's medium overnight, chondrocytes were treated with or without
TNF-α (30 ng/ml) for 2 h. Densitometric analyses of the bands were
conducted by computerized laser densitometry and normalized against
GAPDH. (A) Protein levels of total Akt, p-Akt and GAPDH were
analyzed using western blot analysis. Values are expressed as the
p-Akt/Akt ratio. *P<0.05, **P<0.01 and ***P<0.001. (B)
Protein levels of caspase-3, Bcl-2 and Bax were analyzed using
western blot analysis. Values are expressed as the Bcl-2/Bax ratio
and the caspase-3/GAPDH ratio. *P<0.05, **P<0.01 and
***P<0.001. p-Akt, phospho-Akt; TNF, tumor necrosis factor;
shLRP1, short hairpin RNA targeting low-density lipoprotein
receptor-related protein 1; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein. |
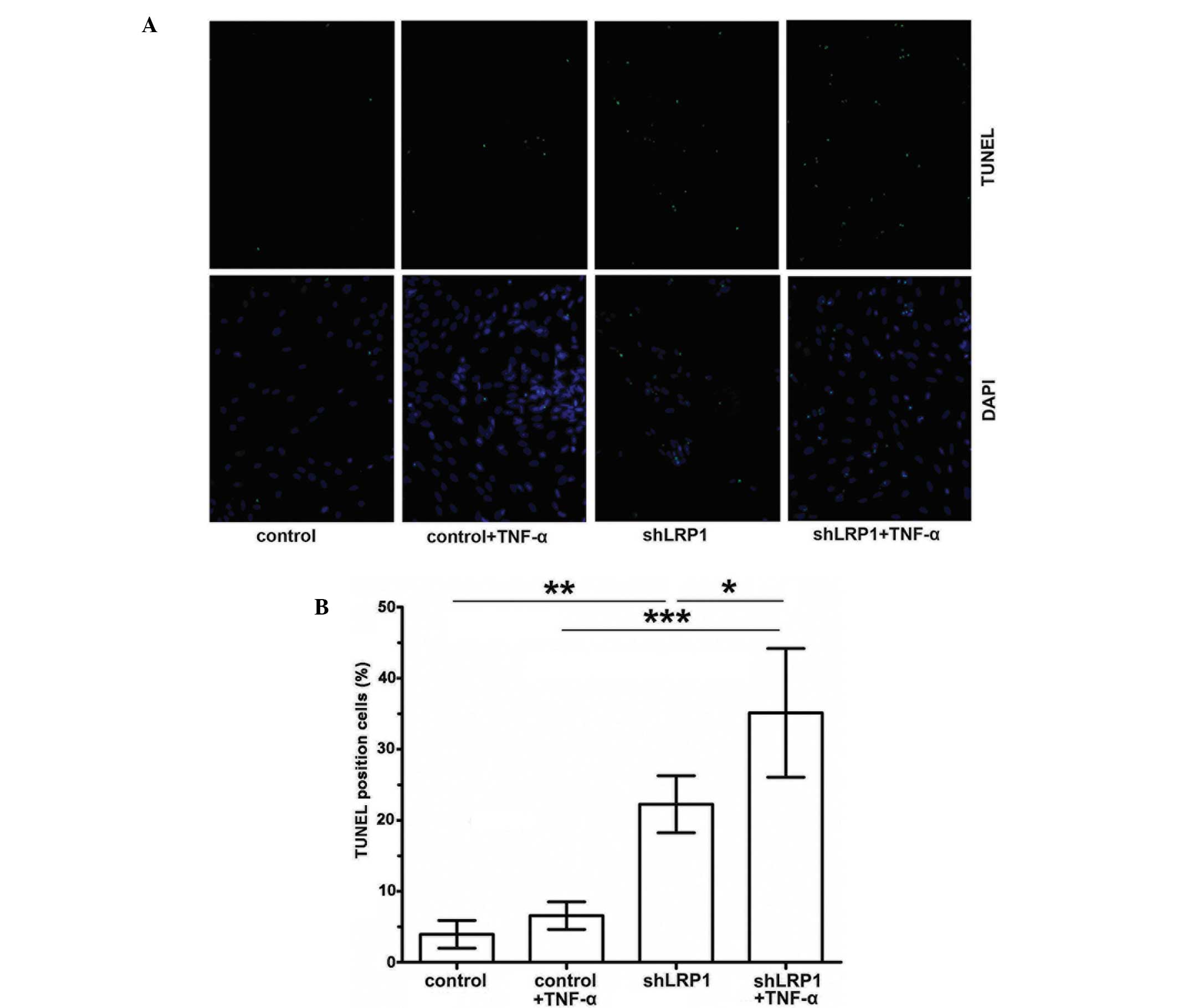
To further confirm the antiapoptotic effect of LRP1
knockdown in vitro, control and shLRP1 chondrocytes were
starved in DMEM overnight, after which the cells were treated with
TNF-α (30 ng/ml) or DMEM for 24 h. The percentage of TUNEL-positive
cells was subsequently determined microscopically. As shown in
Fig. 5A and B, the proportion of
apoptotic cells increased significantly in the shLRP1 chondrocytes
(control, 3±0.96%; shLRP1 + TNF-α, 35.11±4.53%; control + TNF-α,
6.55±0.96%; shLRP1, 21.25±2.01%).
Discussion
Low-density lipoprotein receptor-related protein 1
(LRP1) is a member of the well-studied family of endocytic
receptors, which are larger than other members of the low-density
lipoprotein receptor gene family, but structurally similar
(7). LRP1 participates in the
recognition and endocytosis of lipoproteins, in addition to
recognizing a variety of non-lipoprotein ligands, including
urokinase and tissue plasminogen activator, to participate in a
range of physiological processes (15). As a function of endocytosis, LRP1
regulates the levels of certain matrix metalloproteinase (MMP)
family members. For example, LRP regulates the catabolism and
internalization of MMP-13 via a specific collagenase-3 receptor
that functions as a primary binding site on cells (16).
As a key mediator of inflammatory signaling pathways
(17), LRP1 is able to affect
TNF-α-induced inflammation. The results of the present study
indicate that LRP1 regulates the expression of MMP-13 via the
IKK/NF-κB pathway, which provides a mechanistic explanation for the
anti-inflammatory activity of LRP1.
In order to further investigate the role of LRP1 in
chondrocytes, cells were transfected with lentivirus vectors
expressing LRP1-targeting shRNA, and treatment with shLRP1 was
observed to markedly reduce the levels of LRP1 expression. In
addition, this treatment increased the base levels of TNFR1 in the
absence of exogenous stimulants (Fig.
2B), which was consistent with the results of a previous study
(11). TNF-α, a key proinflammatory
cytokine whose levels are increased in the synovial fluid of
patients with arthritis, has been shown to induce the expression of
MMP-13 (4,18).
The results of the present study indicate that the
levels of phosphorylated NF-κB p65 increase in LRP1-knockdown
chondrocytes in response to TNF-α, which is also accompanied with
increased expression of iNOS and MMP-13. This proposed IKK-NF-κB
pathway mechanism is further supported by the attenuation of TNF-α
induction in chondrocytes by the application of NF-κB inhibitors,
such as Bay 11–7082, which prevents the phosphorylation of the
NF-κB inhibitor, IκB (4). Nitric
oxide (NO) is known to be a crucial inflammatory mediator in the
pathogenesis of OA (19). OA
cartilage generates a large quantity of NO, high levels of which
have been detected in the synovial fluid and serum of patients with
OA (20).
Knockdown of LRP1 was observed to increase the
cell-surface levels of TNFR1. By regulating the cell-surface levels
of TNFR1, LRP1 can control the activity of cell signaling factors
downstream of TNF-α. Knockdown of LRP1 was demonstrated to increase
the activation of the IKK-NF-κB pathway, stimulated by TNF-α, which
was accompanied by an increase in the protein and mRNA expression
levels of MMP-13. These results demonstrate the partial inhibition
of MMP-13 by Bay 11–7082, indicating that LRP-1 is able to inhibit
the IKK-NF-κB pathway, which may be involved in the response of
MMP-13 expression to TNF-α. MMP-13 is a key enzyme involved in
cartilage degradation, which is able to degrade type II
collagen.
In chondrocytes, NF-κB signaling pathways
predominate in the regulation of IL-1 and TNF-α-induced gene
expression. Furthermore, NF-κB pathways are involved in the
inhibition of COL2A1 expression by the aforementioned cytokines
(21–24). As demonstrated by the ELISA results,
extracellular levels of COL2A1 were significantly downregulated in
the shLRP1 chondrocytes treated with TNF-α, which indicated that
LRP1 may indirectly increase the expression of COL2A1.
MMP-13-specific type II collagen cleavage products have been
immunolocalized in OA cartilage (25,26), and
an increase in MMP-13 expression and the suppression of COL2A1 has
been observed in knee joint cartilage exhibiting OA-like changes
(27–29). Thus, the upregulation of MMP-13
expression may contribute to the enhanced rate of cell apoptosis
observed in LRP1-knockdown chondrocytes. Furthermore, LRP1
expresses pro-survival factors in a number of cell types, including
macrophages, neurons and cancer cells (30–32).
Consistent with this hypothesis, LRP1 may decelerate OA progression
and inhibit apoptosis in chondrocytes.
Since various cell types are able to activate the
NF-κB pathway in response to TNF-α via TNFR1, the activation of
NF-κB not only serves a crucial function in inhibiting apoptosis by
inducing the upregulation of key antiapoptotic proteins, such as
Bcl-2, Bcl-extra large and X-linked inhibitor of apoptosis protein
(33,34), but may also perform a proapoptotic
function (35,36). The results of the present study
demonstrate that LRP1-knockdown chondrocytes exhibit increased
susceptibility to cell death in response to TNF-α, as indicated by
the TUNEL assay and the nucleic morphological alterations of
apoptotic chondrocytes stained with 4′,6-diamidino-2-phenylindole
(Fig. 5A). Furthermore, the present
results identify a potential proapoptotic pathway by which
knockdown of LRP1 was able to reduce p-Akt and Bcl-2 expression,
while enhancing the Bax/Bcl-2 ratio and caspase-3 activation. A
possible explanation may be that TNF-α-induced apoptosis is
mediated primarily through the activation of the type I receptor,
which is elevated in LRP1-knockdown chondrocytes. However, p-Akt is
a central regulator of cellular processes in the cytosol, one of
its primary roles is to function indirectly as an antiapoptotic
protein (37). Knockdown of LRP1
expression was shown to increase the levels of caspase-3 and Bax.
Active caspases serve as vital mediators in the induction of
apoptosis. Furthermore, caspase-3 is a downstream regulator of a
variety of proteins, including Bcl-2 family proteins (30,31).
Certain members of the Bcl-2 family, including Bcl-2, are able to
inhibit TNF-induced apoptosis (32).
Bcl-2 and Bax have been identified as key factors influencing cell
susceptibility to apoptosis (31).
In the present study, LRP1 knockdown chondrocytes exhibited reduced
expression levels of Bcl-2 (in particular Bcl-2/Bax ratio), which
indicates that LRP1 is able to induce cellular proliferation and
exert antiapoptotic effects.
In summary, the present study demonstrated a novel
function of LRP1 as an important mediator of TNF-α signaling,
suppressing inflammation and apoptosis in articular chondrocytes.
The inhibitory effects of LRP1 on apoptosis may be partially
derived from the capacity of LRP1 to inhibit TNF-α-induced
apoptosis. Furthermore, the present results indicate that LRP1
exerts a protective effect in cartilage chondrocytes exposed to
TNF-α. However, the precise mechanism underlying LRP1 regulation of
the cross-talk between other key pathways remains unclear, and
further study is required.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 61308110) and by
a faculty research from Wuhan University College of Medicine.
References
|
1
|
Mengshol JA, Vincenti MP, Coon CI,
Barchowsky A and Brinckerhoff CE: Interleukin-1 induction of
collagenase3 (matrix metalloproteinase 13) gene expression in
chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear
factor kappaB: Differential regulation of collagenase 1 and
collagenase 3. Arthritis Rheum. 43:801–811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhong Y, Yu W, Feng J, Fan Z and Li J:
Curcumin suppresses tumor necrosis factor-α-induced matrix
metalloproteinase-2 expression and activity in rat vascular smooth
muscle cells via the NF-κB pathway. Exp Ther Med. 7:1653–1658.
2014.PubMed/NCBI
|
|
3
|
Ling H and Recklies AD: The chitinase
3-like protein human cartilage glycoprotein 39 inhibits cellular
responses to the inflammatory cytokines interleukin-1 and tumour
necrosis factor-alpha. Biochem J. 380:651–659. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liacini A, Sylvester J, Li WQ, Huang W,
Dehnade F, Ahmad M and Zafarullah M: Induction of matrix
metalloproteinase-13 gene expression by TNF-α is mediated by MAP
kinases, AP-1 and NF-κB transcription factors in articular
chondrocytes. Exp Cell Res. 288:208–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campana WM, Li X, Dragojlovic N, Janes J,
Gaultier A and Gonias SL: The low-density lipoprotein
receptor-related protein is a pro-survival receptor in Schwann
cells: Possible implications in peripheral nerve injury. J
Neurosci. 26:11197–11207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zurhove K, Nakajima C, Herz J, Bock HH and
May P: Gamma-secretase limits the inflammatory response through the
processing of LRP-1. Sci Signal. 1:ra152008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herz J and Strickland DK: LRP: A
multifunctional scavenger and signaling receptor. J Clin Invest.
108:779–784. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Kerkhof P, Lee J, McCormick L,
Tetrault E, Lu W, Schoenfish M, Oorschot V, Strous GJ, Klumperman J
and Bu G: Sorting nexin 17 facilitates LRP recycling in the early
endosome. EMBO J. 24:2851–2861. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Herz J: The LDL receptor gene family:
(un)expected signal transducers in the brain. Neuron. 29:571–581.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Overton CD, Yancey PG, Major AS, Linton MF
and Fazio S: Deletion of macrophage LDL receptor-related protein
increases atherogenesis in the mouse. Circ Res. 100:670–677. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gaultier A, Arandjelovic S, Niessen S,
Overton CD, Linton MF, Fazio S, Campana WM, Cravatt BF III and
Gonias SL: Regulation of tumor necrosis factor receptor-1 and the
IKK-NF-κB pathway by LDL receptor-related protein explains the
antiinflammatory activity of this receptor. Blood. 111:5316–5325.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gaultier A, Arandjelovic S, Li X, et al: A
shed form of LDL receptor-related protein-1 regulates peripheral
nerve injury and neuropathic pain in rodents. J Clin Invest.
118:161–172. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu DN, Yang PY, Ku MC, Chu CH, Lim AY and
Hwang MH: Isolation and cultivation of human articular
chondrocytes. Kaohsiung J Med Sci. 18:113–120. 2002.PubMed/NCBI
|
|
14
|
Hermann C, Assmus B, Urbich C, Zeiher AM
and Dimmeler S: Insulin-mediated stimulation of protein kinase Akt:
A potent survival signaling cascade for endothelial cells.
Arterioscler Thromb Vasc Biol. 20:402–409. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gaultier A, Simon G, Niessen S, Dix M,
Takimoto S, Cravatt BF III and Gonias SL: LDL receptor-related
protein 1 regulates the abundance of diverse cell-signaling
proteins in the plasma membrane proteome. J Proteome Res.
9:6689–6695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barmina OY, Walling HW, Fiacco GJ, Freije
JM, López-Otín C, Jeffrey JJ and Partridge NC: Collagenase-3 binds
to a specific receptor and requires the low density lipoprotein
receptor-related protein for internalization. J Biol Chem.
274:30087–30093. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raggatt LJ, Jefcoat SC Jr, Choudhury I,
Williams S, Tiku M and Partridge NC: Matrix metalloproteinase-13
influences ERK signalling in articular rabbit chondrocytes.
Osteoarthritis Cartilage. 14:680–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Manicourt DH, Poilvache P, Van Egeren A,
Devogelaer JP, Lenz ME and Thonar EJ: Synovial fluid levels of
tumor necrosis factor alpha and oncostatin M correlate with levels
of markers of the degradation of crosslinked collagen and cartilage
aggrecan in rheumatoid arthritis but not in osteoarthritis.
Arthritis Rheum. 43:281–288. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Henrotin Y, Kurz B and Aigner T: Oxygen
and reactive oxygen species in cartilage degradation: Friends or
foes? Osteoarthritis Cartilage. 13:643–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Henrotin Y and Kurz B: Antioxidant to
treat osteoarthritis: Dream or reality? Curr Drug Targets.
8:347–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Séguin CA and Bernier SM: TNFalpha
suppresses link protein and type II collagen expression in
chondrocytes: Role of MEK1/2 and NF-kappaB signaling pathways. J
Cell Physiol. 197:356–369. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Robbins JR, Thomas B, Tan L, Choy B,
Arbiser JL, Berenbaum F and Goldring MB: Immortalized human adult
articular chondrocytes maintain cartilage-specific phenotype and
responses to interleukin-1beta. Arthritis Rheum. 43:2189–2201.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murakami S, Lefebvre V and de Crombrugghe
B: Potent inhibition of the master chondrogenic factor Sox9 gene by
interleukin-1 and tumor necrosis factor-alpha. J Biol Chem.
275:3687–3692. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sitcheran R, Cogswell PC and Baldwin AS
Jr: NF-kappaB mediates inhibition of mesenchymal cell
differentiation through a posttranscriptional gene silencing
mechanism. Genes Dev. 17:2368–2373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tetlow LC, Adlam DJ and Woolley DE: Matrix
metalloproteinase and proinflammatory cytokine production by
chondrocytes of human osteoarthritic cartilage: Associations with
degenerative changes. Arthritis Rheum. 44:585–594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu W, Billinghurst RC, Pidoux I, Antoniou
J, Zukor D, Tanzer M and Poole AR: Sites of collagenase cleavage
and denaturation of type II collagen in aging and osteoarthritic
articular cartilage and their relationship to the distribution of
matrix metalloproteinase 1 and matrix metalloproteinase 13.
Arthritis Rheum. 46:2087–2094. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neuhold LA, Killar L, Zhao W, Sung ML,
Warner L, Kulik J, et al: Postnatal expression in hyaline cartilage
of constitutively active human collagenase-3 (MMP-13) induces
osteoarthritis in mice. J Clin Invest. 107:35–44. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aigner T, Vornehm SI, Zeiler G, Dudhia J,
von der Mark K and Bayliss MT: Suppression of cartilage matrix gene
expression in upper zone chondrocytes of osteoarthritic cartilage.
Arthritis Rheum. 40:562–569. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aigner T, Zien A, Gehrsitz A, Gebhard PM
and McKenna L: Anabolic and catabolic gene expression pattern
analysis in normal versus osteoarthritic cartilage using
complementary DNA-array technology. Arthritis Rheum. 44:2777–2789.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cui N, Li S, Zhao X, Zhang T, Zhang C, Yu
L, Zhu Z and Xie K: Expression of Bcl-2, Bax and Caspase-3 in nerve
tissues of rats chronically exposed to 2,5-hexanedione. Neurochem
Res. 32:1566–1572. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Postolow F, Fediuk J, Nolette N, Hinton M
and Dakshinamurti S: Hypoxia and nitric oxide exposure promote
apoptotic signaling in contractile pulmonary arterial smooth muscle
but not in pulmonary epithelium. Pediatr Pulmonol. 46:1194–1208.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rath PC and Aggarwal BB: TNF-induced
signaling in apoptosis. J Clin Immunol. 19:350–364. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Graham B and Gibson SB: The two faces of
NFkappaB in cell survival responses. Cell Cycle. 4:1342–1345. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luo JJ, Li CY, Liu S, Yu W, Tang SY, Cai
HL and Zhang Y: Overexpression of Helicobacter pylori VacA
N-terminal fragment induces proinflammatory cytokine expression and
apoptosis in human monocytic cell line through activation of NF-κB.
Can J Microbiol. 59:523–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Xiao Q, Zhao K and Gao Y: Ghrelin
inhibits high glucose-induced PC12 cell apoptosis by regulating
TLR4/NF-κB pathway. Inflammation. 36:1286–1294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Swaminathan JK, Khan M, Mohan IK,
Selvendiran K, Niranjali Devaraj S, Rivera BK and Kuppusamy P:
Cardioprotective properties of Crataegus oxycantha extract against
ischemia-reperfusion injury. Phytomedicine. 17:744–752. 2010.
View Article : Google Scholar : PubMed/NCBI
|