Introduction
Vinorelbine (VIN) is a semi-synthetic vinca alkaloid
with an antitumor activity that is associated with its ability to
induce the depolymerization of microtubules and disrupt the mitotic
spindle apparatus (1). At present,
VIN is one of the most active agents used in the treatment of
non-small cell lung cancer and other solid tumors (2–4). VIN is
typically administered through a peripheral vein (2–4);
however, the drug is a moderate vesicant and causes local venous
toxicity. The incidence of VIN-induced local venous toxicity has
been reported to be ~30% (5,6). Although infusion phlebitis is common,
its pathogenesis is not fully understood; however, it has been
suggested that the pathogenesis is associated with chemical
irritation of the endothelial cells, leading to sterile
inflammation and thrombosis (7,8).
Toll-like receptors (TLRs) are a class of pattern
recognition receptors that are important sensors of pathogen
invasion. To date, 13 members of the TLR family have been
identified in mammals. These TLRs respond to different
microbe-associated molecular patterns, including viral and
bacterial nucleic acids, lipopolysaccharide (LPS), lipoteichoic
acids and flagellin (9), although
they can also bind endogenously generated ligands, such as heat
shock proteins (10,11), surfactant protein A18 (12), extracellular matrix components
(13) and high-mobility group box-1
(14,15). All members of the TLR family, with
the exception of TLR3, signal through an adaptor protein known as
MyD88, which is responsible for stimulating a cascade of events
that leads to the activation of the transcription factor nuclear
factor-κB (NF-κB) (16).
NF-κB is an important regulator of various genes
involved in immune and inflammatory responses and is linked to
inflammatory and growth responses. TLR4 is known to induce NF-κB
activation; however, it is not yet clear whether VIN induces the
activation of the TLR4/NF-κB pathway in human umbilical vein
endothelial cells (HUVECs). Furthermore, the association between
the activation of NF-κB and TLR4 caused by VIN has yet to be
revealed. TLR4 is known to participate in the selective relay of a
variety of signals from the membrane to transducers of cell
activation and gene expression (16). We postulated that TLR4 was a
potential contributing factor in the pathogenesis of VIN-induced
vascular endothelial injury. The aim of the present study,
therefore, was to examine the effects of VIN on the expression of
TLR4 and the transcription factor NF-κB.
Materials and methods
HUVEC preparation and culture
HUVECs were prepared using human umbilical veins
from umbilical cords recovered with the written informed consent of
the parents. Cells were cultured in basal EBM™-2 basal medium
supplemented with growth factor (Lonza Group, Basel, Switzerland)
at 37°C and under 5% CO2. EBM-2 basal medium was
supplemented with an EGM™-2 MV SingleQuot™ kit, also from Lonza
Group. Cells from between passages two and five were used.
Exposure of HUVECs to VIN
Cells at a density of 5×104 cells/ml were
seeded in 25-cm2 flasks or 24-well plates. Upon reaching
90–95% confluence, the cells were washed twice with
phosphate-buffered saline (PBS) and fed VIN solution at a final
concentration of 0.05 mg/l in the flasks or wells. Untreated
control cells were fed medium without VIN. After 1 h of incubation
with VIN, the cells were washed twice with PBS, re-fed medium
without VIN and then grown for 0, 6 or 12 h. The apoptotic cells
were quantified by the Annexin V-PE/7-AAD apoptosis detection kit I
(BD Biosciences, Franklin Lakes, NJ, USA). These cells were then
washed twice with PBS and harvested for quantitative polymerase
chain reaction (qPCR), western blot and immunofluorescence
analyses. Experiments were repeated at least three times.
RNA isolation
Cells (106) were suspended in 1 ml
TRIzol™ reagent (Invitrogen Life Technologies, Carlsbad, CA, USA)
in a 2-ml screw-cap microtube. After 5 min at room temperature, 0.2
ml chloroform was added and the tube was agitated for 15 sec. The
tube was then allowed to sit at room temperature for 10 min, prior
to centrifugation at 12,000 × g and 4°C for 10 min and the
transferral of the supernatant to a Qiagen RNeasy® Mini kit column
(Qiagen, Crawley, UK). Total RNA was extracted according to the
manufacturer's instructions. RNA samples were stored at −80°C until
use.
qPCR
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen Life Technologies) following the manufacturer's
instructions. The cDNA was generated with an oligo (dt) primer
(Invitrogen Life Technologies), according to the manufacturer's
instructions. qPCR was performed using the ABI Prism® 7500 Sequence
Detection System (Applied Biosystems, Branchburg, NJ, USA) with ABI
PRISM® 7000 SDS software (Applied Biosystems). PCR was carried out
with SYBR® Green PCR Master Mix (Invitrogen Life Technologies)
using 1 µl cDNA in a 25-µl final reaction mixture. The reaction
mixture was heated initially at 94°C for 2 min, followed by 35
cycles of denaturation at 94°C for 30 sec and annealing/extension
at 60°C for 30 sec. Data were analyzed using the 2−∆∆CT
method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was
utilized for endogenous quantity control. The primers used were as
follows: TLR4 forward, 5′-AAGCCGAAAGGTGATTGTTG-3′ and reverse,
5′-CTGAGCAGGGTCTTCTCCAC-3′; GAPDH forward, 5′-GGGAAACTGTGGCGTGAT-3′
and reverse, 5′-GAGTGGGTGTCGCTGTTGA-3′.
Western blot analysis
Logarithmic growth phase HUVECs were treated with
the test compounds or with blank vehicle for the specified lengths
of time and then lysed in cell lysis buffer on ice for 40 min.
Soluble protein was recovered following centrifugation at 4°C at
10,000 × g for 30 min. Samples were assayed for protein
concentration using the bicinchoninic acid assay. The samples were
separated on 10% SDS-PAGE gels and then transferred to
nitrocellulose membranes. The membranes were incubated with a
polyclonal rabbit anti-human TLR4 antibody (1:500 dilution; cat.
no. 2246, Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight and then incubated with peroxidase-labeled goat
anti-rabbit secondary antibody (1:5,000; cat. no. A0545,
Sigma-Aldrich, St. Louis, MO, USA). Mouse polyclonal β-actin
primary antibody was used as a control (1:1,000 dilution; cat. no.
612657, BD Biosciences). Immunoreactive bands were visualized using
enhanced chemiluminescence reagents. The protein levels were
quantified using Quantity One® software (Bio-Rad, Hercules, CA,
USA).
Immunofluorescence and confocal
analysis
Cells on coverslips were fixed in 4%
paraformaldehyde for 15 min at room temperature. Following three
washes with PBS, the cells were permeabilized with ice-cold
methanol for 10 min at −20°C. The cells were then rinsed three
times with PBS and incubated for 1 h at room temperature in the
dark in a PBS solution containing 2% (wt/v) bovine serum albumin
(Merck Millipore, Darmstadt, Germany). Following incubation, the
cells were rinsed three more times in PBS and incubated overnight
with primary antibodies at 4°C. The slides were washed a further
three times and incubated with Sytox® Green Nucleic Acid Stain
(cat. no. S7020; 1:10,000 dilution in Milli-Q water; Invitrogen
Life Technologies) at room temperature in the dark for 1 h.
Following incubation, the cells were washed three times with PBS.
The nuclear dye DAPI was applied and the cells were incubated for
15 min. Following a final wash, the coverslips were mounted in
Fluoromount™ (Vector Laboratories, Inc., Burlingame, CA, USA) and
sealed with nail varnish. Stained slides were examined using a
Confocal Laser Scanning Biological Microscope (Olympus Fluoview
FV1000; Olympus Optical Co., Ltd., Tokyo, Japan) equipped with an
Olympus IX70 camera and recorded as a high-resolution layer.
Statistical analysis
Statistical analysis was performed using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). Results are expressed the
mean ± standard deviation. Experimental data were analyzed using
one-way analysis of variance followed by Tukey's multiple range
test for significance. P<0.05 was considered to indicate a
statistically significant difference.
Results
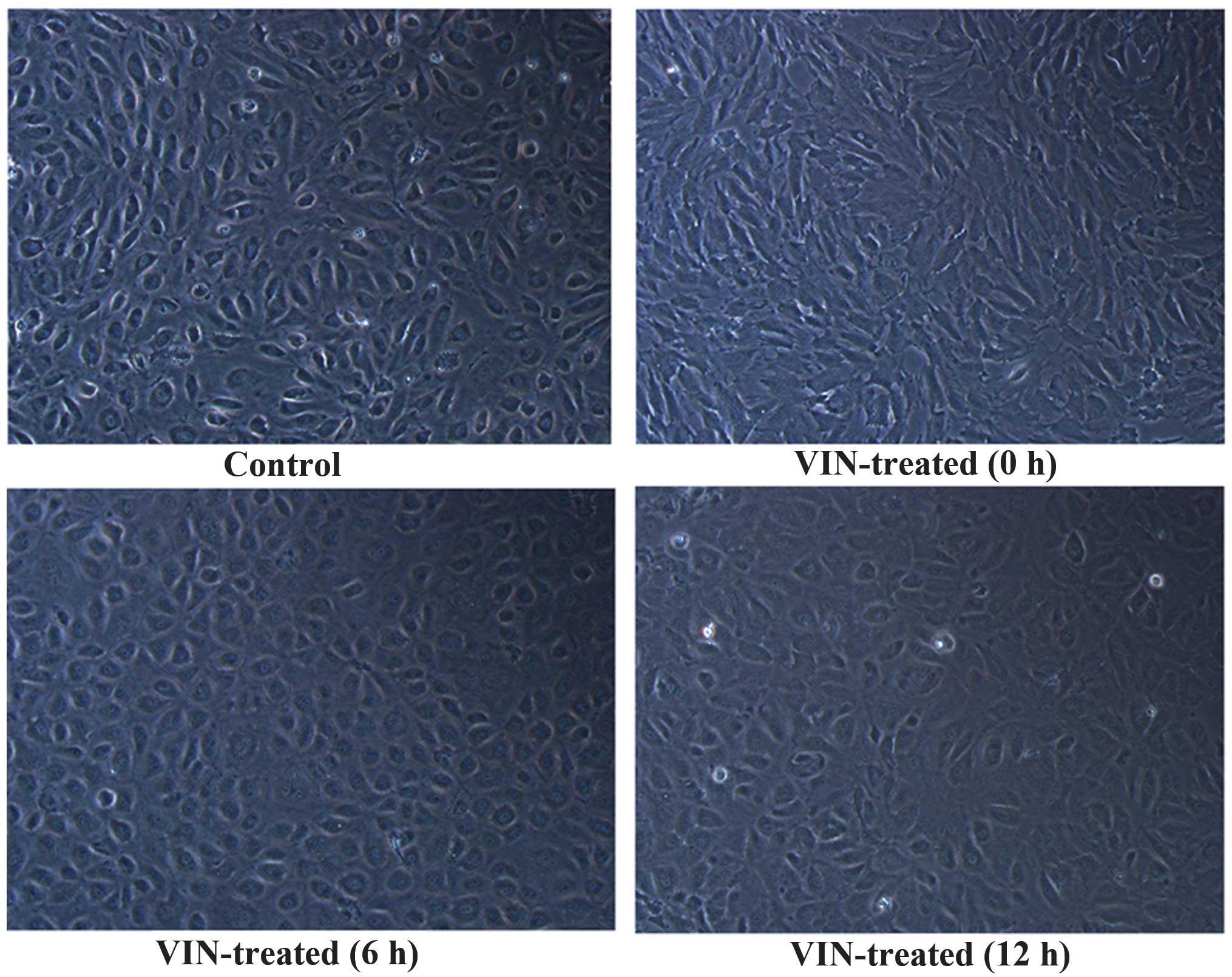
Morphological changes in the
HUVECs
HUVECs in the vehicle-treated group showed adherent
dermoid growth (Fig. 1), a
paving-stone arrangement, spindle-shaped cell morphology and nuclei
with abundant cytoplasm. The VIN-treated cells were stretched,
extended, irregular and disordered. Once the VIN had been washed
away, the cells were cultured for a further 6 and 12 h, during
which cell morphology gradually returned to normal.
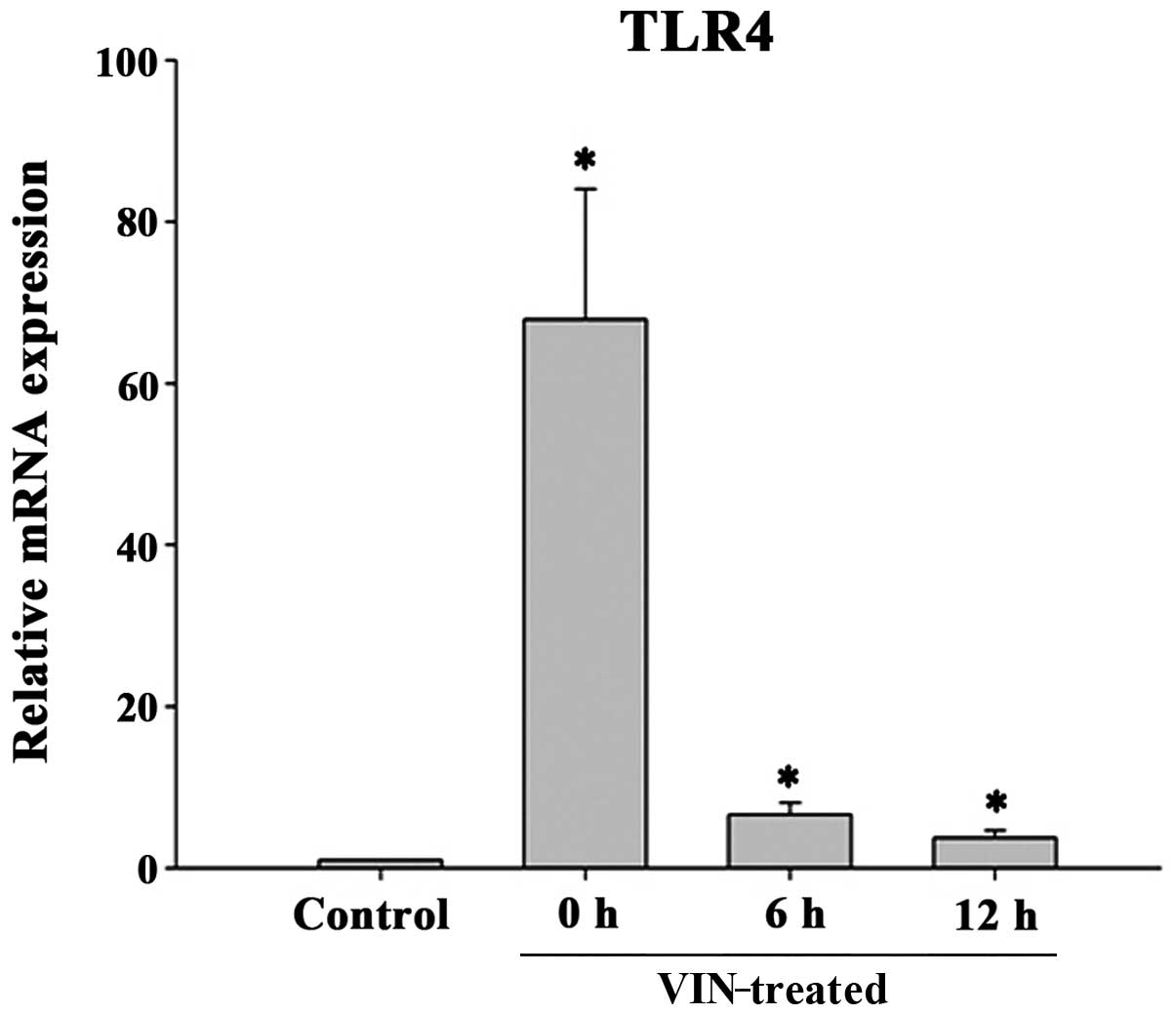
TLR4 mRNA expression profile in HUVECs
exposed to VIN
qPCR was performed to verify the changes in TLR4
mRNA expression in the HUVECs. As shown in Fig. 2, the levels of TLR4 mRNA expression
were significantly higher in the VIN-treated HUVECs than those in
the vehicle-treated group (P<0.05). The effects of VIN peaked 1
h after initial exposure.
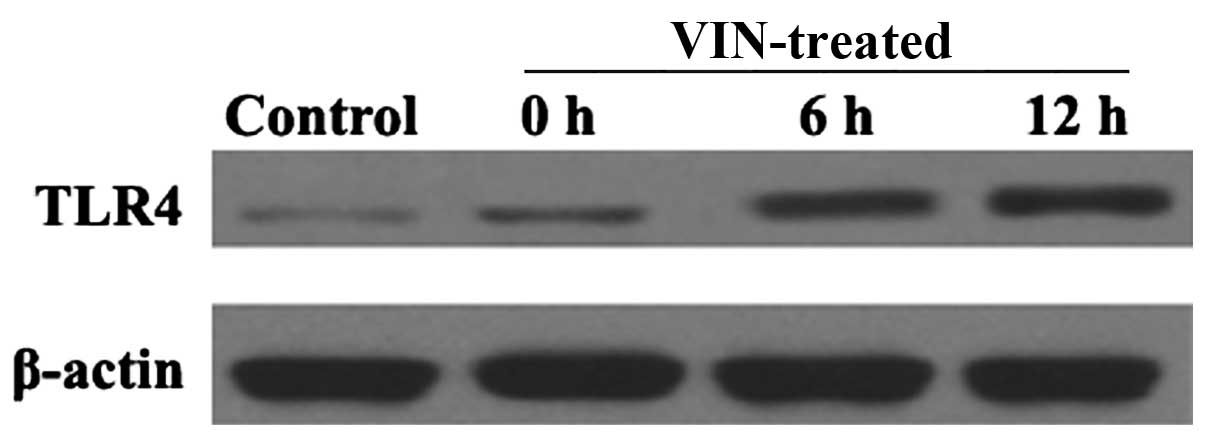
TLR4 protein expression profile of
HUVECs exposed to VIN
Western blotting was performed to assess the
relative levels of TLR4 protein expression in the HUVECs and
determine the effects of VIN treatment. As shown in Fig. 3, the level of TLR4 protein expression
in the VIN-treated groups was significantly higher than that in the
vehicle-treated group.
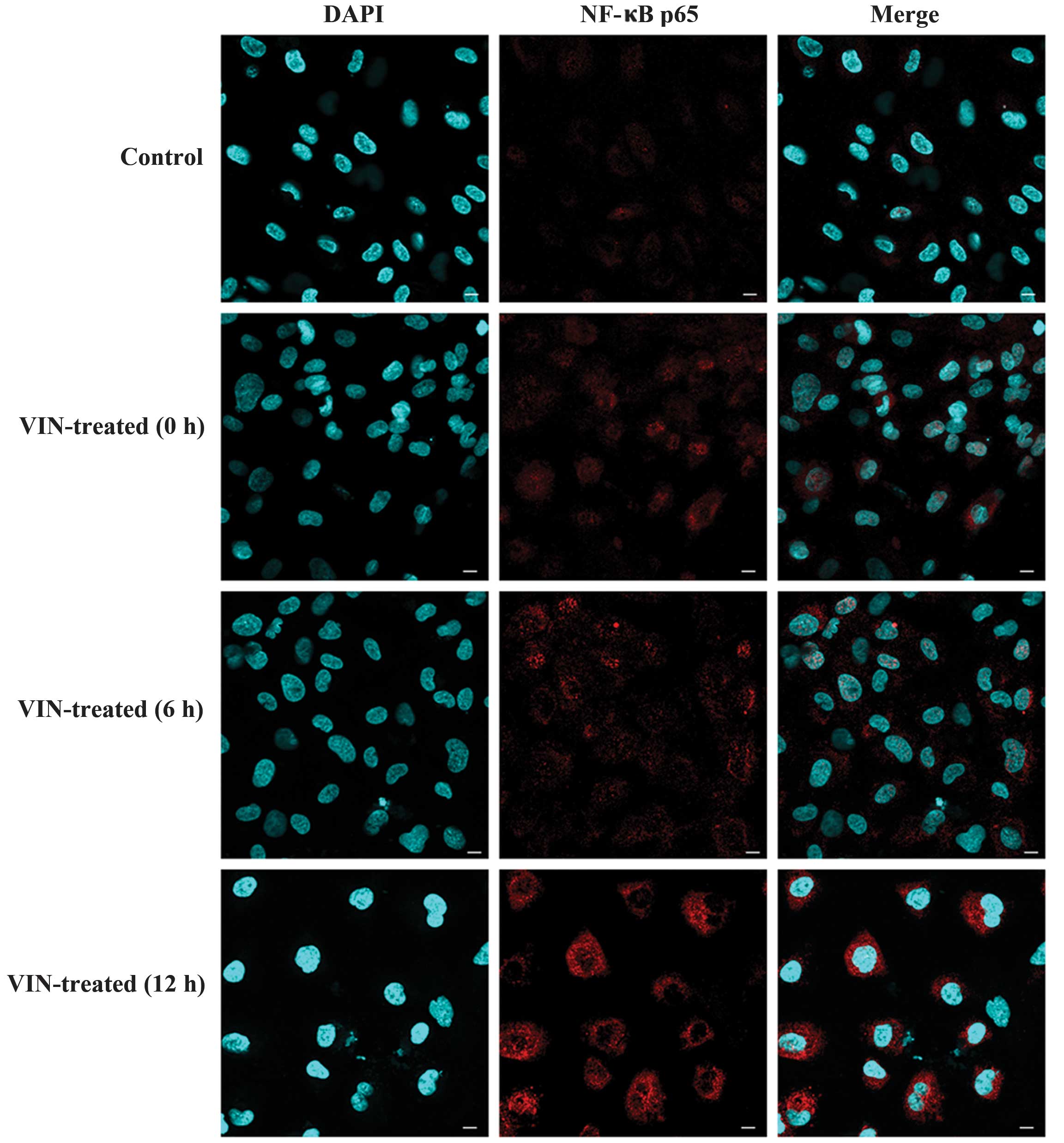
Effects of VIN on the activation of
NF-κB p65
Given the crucial role attributed to VIN in the
induction of vascular endothelial damage, the next focus of the
study was on NF-κB signaling. As shown in Fig. 4, NF-κB activation, as indicated by
NF-κB phosphorylation, was assessed at different times and became
undetectable 20 h after treatment (data not shown). As expected,
nuclear translocation of p65 occurred in the HUVECs treated with
VIN, and NF-κB activation occurred within a narrow window in the
HUVECs subjected to vascular endothelial injury.
Effects of VIN on apoptosis
VIN-induced apoptosis was quantified using Annexin
V/7-AAD. The results indicated that the apoptotic rate of the
HUVECs was increased in response to VIN, in a dose-dependent manner
(data not shown). This result suggests that the increased apoptosis
of the HUVECs had a close association with the decreased expression
levels of TLR4, as caused by treatment with VIN.
Discussion
Vascular endothelial cells adhere to the vascular
walls, forming an integral part of the blood vessels and exhibiting
a variety of immunological functions. The dysfunction of
endothelial cell apoptosis has been observed in human diseases and
experimental models, including atherosclerosis,
ischemia-reperfusion and sepsis (17–19). VIN
is a moderate vesicant that has been known to cause local venous
toxicity (3,6). Although the pathogenesis of VIN has
been little reported, it has previously been suggested that
improper endothelial cell apoptosis could be responsible for the
local venous toxicity caused by the VIN (19). Thus, primary HUVECs were selected for
the present study in order to reveal their roles in the protection
against vascular endothelial injury.
The morphology experiments suggested that VIN
significantly changed the morphology of the HUVECs, causing them to
stretch, extend and become irregular and disordered. The results
indicated that the VIN-induced HUVEC injury involved disruption of
the morphological structure of the cell and that the removal of the
stimuli allowed the cells to self-regulate and return to their
normal state. Furthermore, consistent with the present findings, it
has previously been shown that VIN can induce apoptotic cell death
in HUVECs (19). Thus, the present
and previous findings have indicated that endothelial cell injury
can contribute to VIN-induced vascular injury.
In a previous study by Frantz et al (20) it was found that TLR4 was
constitutively expressed in cardiac myocytes and that an
upregulation of TLR4 expression was present in the hearts of humans
with cardiomyopathies and rodents with experimental cardiac
dysfunction. Furthermore, in cultured vascular endothelial cells
TLR4 expression has been shown to increase from baseline to high
levels following stimulation with proinflammatory cytokines
(21,22).
Several studies have suggested that, when tissue is
damaged, a variety of endogenous ligands are produced, which can
stimulate and promote the expression of TLR4 (3,13).
Vascular endothelial cell apoptosis and injury are involved in the
formation of phlebitis (19,21). This may indicate that the endogenous
ligands are released following endothelial cell injury, possibly
leading to the upregulation of TLR4 in the endothelial cells
(19,21). The qPCR and western blot analysis
showed that TLR4 mRNA and protein levels were significantly higher
in the VIN-treated cells than those in the control group. These
results indicate that TLR4 is involved in VIN-induced vascular
endothelial injury by affecting gene transcription and protein
synthesis. This result is consistent with the finding of previous
studies that VIN stimulation can lead to the upregulation of TLR4
in endothelial cells (19,20,22).
NF-κB plays a central role in inflammation through
its ability to induce the transcription of proinflammatory genes
(23). In resting cells, NF-κB is
localized in the cytoplasm due to the binding of inhibitor of κB
(IκB) proteins (23). The induction
of NF-κB activation by TLR4 is dependent upon the phosphorylation
of IκBα mediated by the IκB kinases IKKα and IKKβ. IκBα
phosphorylation stimulates its ubiquitination and proteasomal
degradation, thus releasing NF-κB and enabling its translocation to
the nucleus, where it can activate the transcription of genes
encoding inflammatory molecules (24).
A previous study suggested that NF-κB may contribute
to endothelial cell injury by affecting inflammatory molecules
(23). As NF-κB p65 is
representative of the NF-κB family (14), the effect of VIN on the activation of
NF-κB p65 was investigated in the present study. The results showed
that VIN promoted NF-κB p65 activation and translocation to the
nucleus, which stimulated the release of a range of inflammatory
factors. The activation of the NF-κB pathway is adapted to the
upregulation of numerous immune and stress response genes, which
terminate the activation (24). The
rates of translocation of NF-κB p65 and the expression level of
TLR4 mRNA initially increased in the VIN-treated cells, then
decreased.
In a study by Frantz et al (20) it was indicated that a ‘basal’ level
of NF-κB activity was necessary for the maintenance of TLR4
expression. Specifically, it was suggested that ammonium
pyrrolidine dithiocarbamate could reproducibly suppress TLR4 mRNA
and protein abundance in the cells of both normal and failing
myocardium, even in the absence of interleukin (IL)-1 and LPS
(20). For this reason, we
hypothesized that the activity of NF-κB plays an important role in
the regulation of TLR4. TLR4 activation can also activate NF-κB,
suggesting that there may be a positive feedback mechanism between
the two. The fact that TLR4 activates the NF-κB pathway has been
known for some time, but the manner in which NF-κB regulates TLR4
expression is not clear, and further investigation is required.
In conclusion, the present study showed that
exposure of HUVECs to 0.05 mg/l VIN upregulates TLR4 mRNA and
protein expression, activating NF-κB p65. These results suggest
that VIN induces HUVEC injury, possibly via the TLR4/NF-κB pathway.
TLR4 is stimulated by VIN and signals to the transcription factor
NF-κB, which regulates the expression of numerous inflammatory
cytokines and chemokines. Our data may provide a novel mechanism
for VIN-induced phlebitis. VIN can injure HUVECs, releasing a
variety of signals associated with danger and death. This in turn
can activate TLR4 and elevate the activity of the NF-κB pathway. To
the best of our knowledge, this is the first report to describe a
role for TLR4 in VIN-induced phlebitis in vitro. In this
way, this study provides novel experimental evidence regarding
potential therapeutic targets for the prevention and treatment of
VIN-induced phlebitis. Further study is required to determine the
specific role of TLR4 in VIN-induced vascular endothelial
injury.
Acknowledgements
The authors would like to thank Professor Hongyan
Dong and Dr Qingyun Wu (Department of Hematology, The Affiliated
Hospital of Xuzhou Medical College, Xuzhou, China) for their
technical assistance.
References
|
1
|
Potier P: The synthesis of Navelbine
prototype of a new series of vinblastine derivatives. Semin Oncol.
16:(2 Suppl 4). 2–4. 1989.PubMed/NCBI
|
|
2
|
Depierre A, Lemarie E, Dabouis G, et al: A
phase II study of Navelbine (vinorelbine) in the treatment of
non-small-cell lung cancer. Am J Clin Oncol. 14:115–119. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fumoleau P, Delgado FM, Delozier T, et al:
Phase II trial of weekly intravenous vinorelbine in first-line
advanced breast cancer chemotherapy. J Clin Oncol. 11:1245–1252.
1993.PubMed/NCBI
|
|
4
|
Devizzi L, Santoro A, Bonfante V, et al:
Vinorelbine: a new promising drug in Hodgkin's disease. Leuk
Lymphoma. 22:409–414. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoh K, Niho S, Goto K, et al: High body
mass index correlates with increased risk of venous irritation by
vinorelbine infusion. J Clin Oncol. 34:206–209. 2004.
|
|
6
|
Yoh K, Niho S, Goto K, et al: Randomized
trial of drip infusion versus bolus injection of vinorelbine for
the control of local venous toxicity. Lung Cancer. 55:337–341.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lewis GB and Hecker JF: Infusion
thrombophlebitis. Br J Anaesth. 57:220–233. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Falchuk KH, Peterson L and McNeil BJ:
Microparticulate-induced phlebitis. Its prevention by in-line
filtration. N Engl J Med. 312:78–82. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyake K: Innate immune sensing of
pathogens and danger signals by cell surface Toll-like receptors.
Semin Immunol. 19:3–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ohashi K, Burkart V, Flohé S and Kolb H:
Cutting edge: heat shock protein 60 is a putative endogenous ligand
of the toll-like receptor-4 complex. J Immunol. 164:558–561. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vabulas RM, Braedel S, Hilf N, et al: The
endoplasmic reticulum-resident heat shock protein Gp96 activates
dendritic cells via the Toll-like receptor 2/4 pathway. J Biol
Chem. 277:20847–20853. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roelofs MF, Boelens WC, Joosten LA, et al:
Identification of small heat shock protein B8 (HSP22) as a novel
TLR4 ligand and potential involvement in the pathogenesis of
rheumatoid arthritis. J Immunol. 176:7021–7027. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guillot L, Balloy V, McCormack FX, et al:
Cutting edge: the immunostimulatory activity of the lung surfactant
protein-A involves Toll-like receptor 4. J Immunol. 168:5989–5992.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rakoff-Nahoum S and Medzhitov R:
Regulation of spontaneous intestinal tumorigenesis through the
adaptor protein MyD88. Science. 317:124–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park JS, Svetkauskaite D, He Q, et al:
Involvement of toll-like receptors 2 and 4 in cellular activation
by high mobility group box 1 protein. J Biol Chem. 279:7370–7377.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kawai T and Akira S: TLR signaling. Semin
Immunol. 19:24–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tricot O, Mallat Z, Heymes C, et al:
Relation between endothelial cell apoptosis and blood flow
direction in human atherosclerotic plaques. Circulation.
101:2450–2453. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scarabelli TM, Stephanou A, Pasini E, et
al: Different signaling pathways induce apoptosis in endothelial
cells and cardiac myocytes during ischemia/reperfusion injury. Circ
Res. 90:745–748. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bannerman DD and Goldblum SE: Mechanisms
of bacterial lipopolysaccharide induced endothelial apoptosis. Am J
Physiol Lung Cell Mol Physiol. 284:L899–L914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Frantz S, Kobzik L, Kim YD, et al: Toll4
(TLR4) expression in cardiac myocytes in normal and failing
myocardium. J Clin Invest. 104:271–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faure E, Equils O, Sieling PA, et al:
Bacterial lipopolysaccharide activates NF-kappaB through toll-like
receptor 4 (TLR-4) in cultured human dermal endothelial cells.
Differential expression of TLR-4 and TLR-2 in endothelial cells. J
Biol Chem. 275:11058–11063. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Faure E, Thomas L, Xu H, et al: Bacterial
lipopolysaccharide and IFN-gamma induce Toll-like receptor 2 and
Toll-like receptor 4 expression in human endothelial cells: role of
NF-kappa B activation. J Immunol. 166:2018–2024. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Winn RK and Harlan JM: The role of
endothelial cell apoptosis in inflammatory and immune diseases. J
Thromb Haemost. 3:1815–1824. 2005. View Article : Google Scholar : PubMed/NCBI
|