Introduction
In 1979, Arakawa et al first described a new
glomerular disease with collagen fibrils in the mesangial matrix
and subendothelial space (1).
Subsequently, in 1990, Ikeda et al proved that the collagen
fibrils in the renal glomerulus were type III collagen fibrils by
immunohistochemical staining (2).
This new glomerular disease was officially named collagen type III
glomerulopathy by Imbasciati et al in 1991 (3). The clinical manifestations of this
disease are proteinuria, edema, hypertension and occasional
progression to end-stage renal disease (4,5). The
etiology and pathogenesis remain elusive. Although the majority of
the initial studies were from Japan, there are currently studies
from around the globe including Europe, South America, North
America and Asia. However, studies from China are rare in the
English literature, and the specific features of Chinese cases are
yet to be summarized. Here, we report a case of collagen type III
glomerulopathy with two differing renal biopsies and review 20
cases in China to investigate the idiographic features of collagen
type III glomerulopathy in China. This study was conducted in
accordance with the declaration of Helsinki, and with the approval
of the Ethics Committee of Puai Hospital, Tongji Medical College,
Huazhong University of Science and Technology (Wuhan, China).
Written informed consent was obtained from the participant.
Case report
In July 2011, a 12-year-old male presented with a
one-year history of edema and proteinuria. The patient was
suffering lower extremity and periorbital edema associated with
hypertension. He had no nail or patella dysplasia and no family
history of renal disease. Laboratory results included urine protein
levels of 2.7 g/24 h (0.028–0.141 g/24 h) and no microscopic
hematuria, hemoglobin levels of 86 g/l (normal level 131–172 g/l),
serum albumin levels of 26.3 g/l (normal level 34–48 g/l), blood
urea nitrogen levels of 7.92 mmol/l (normal level 2.9–8.2 mmol/l),
creatinine levels of 122.8 µmol/l (normal level 62–115 µmol/l),
elevated uric acid levels of 457.1 µmol/l (normal level 208–428
µmol/l), C3 levels of 0.61 g/l (normal level 0.79–1.52 g/l) and
normal C4 levels. Screening for auto-antibodies was negative. The
first renal biopsy was performed on July 26, 2011, but only one
extremely small specimen was taken as the patient was extremely
anxious and the patient's blood pressure was elevated during the
procedure. The specimen was processed by immunofluorescence
microscopy and electronic microscopy scanning; however, paraffin
sections revealed no glomeruli. Thus, the frozen sections were
embedded for light microscopy. From the immunofluorescence
microscopy, four glomeruli were observed. IgM was positive in the
mesangium, and IgA, IgG, C3 and C1q were weakly positive in the
mesangium. Under light microscopy, the glomeruli exhibited
segmentation and mild mesangial hypercellularity. Under electronic
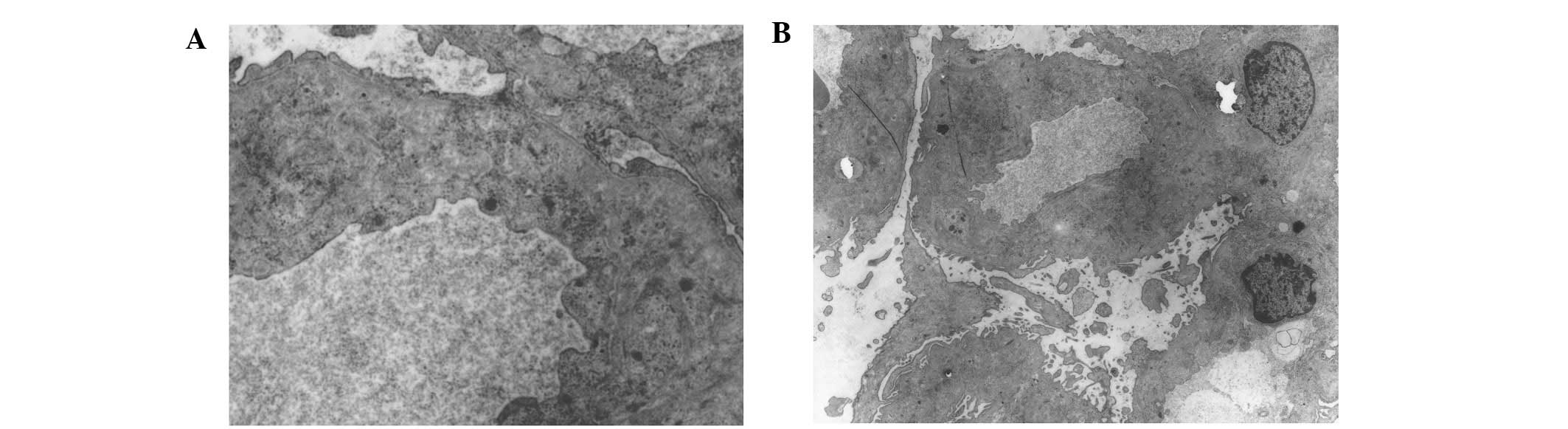
microscopy scanning, one glomerulus demonstrated no electron-dense
or collagen fibril deposits in the mesangial matrix and
subendothelial space (Fig. 1). After
being treated with ACE inhibitor for one year, the edema and
hypertension improved but the proteinuria and anemia persisted. The
patient was readmitted to the hospital on July 19, 2012. Laboratory
results revealed urine protein levels of 1.94 g/24 h without
microscopic hematuria, low hemoglobin levels of 80 g/l, iron panel
consistent with iron deficiency, blood urea nitrogen levels of 7.35
mmol/l, creatinine levels of 94.1 µmol/l, uric acid levels of 436.6
µmol/l and C3 levels remained low at 0.468 g/l. A second renal
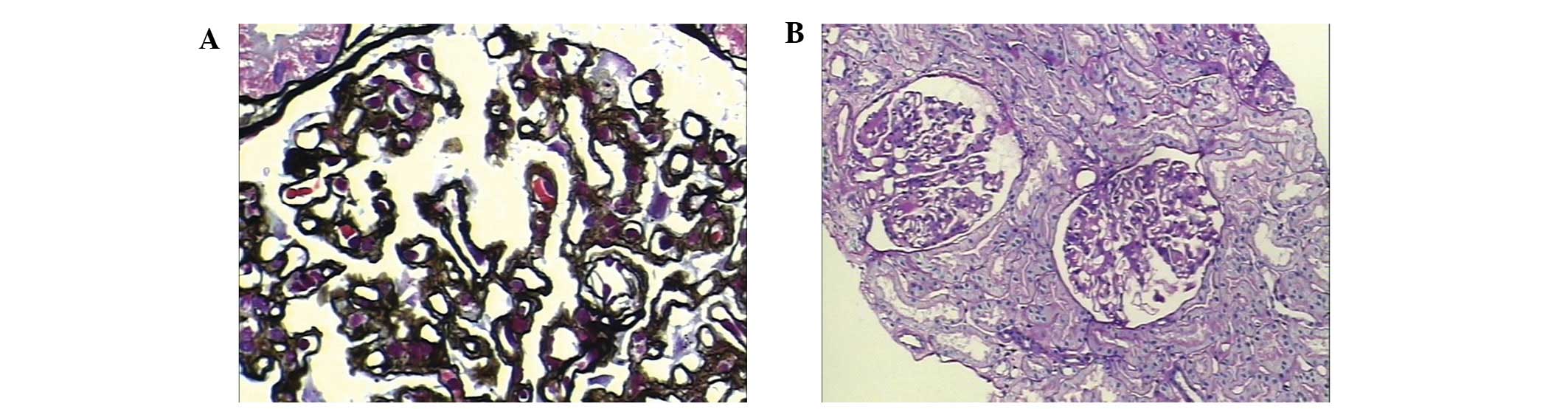
biopsy was then performed on August 2, 2012. Under light
microscopy, there were 24 glomeruli, of which one was completely
sclerotic. The remainder presented mild mesangial hypercellularity.
The basal membrane was crumpled, and there was fake double-tracks
in certain segments. No significant immune complex deposition was
identified by Masson staining. Foci of interstitial fibrosis and
arteriolar hyaline deposits were identified (Fig. 2). By immunofluorescence microscopy,
there were four glomeruli. IgA, IgM, IgG and complement C3 were all
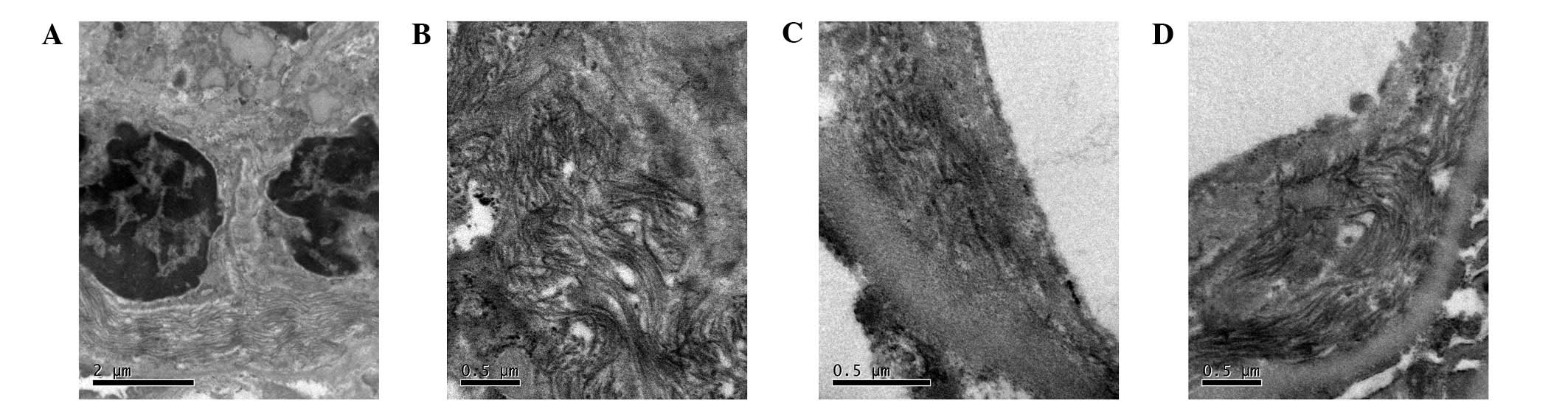
negative. By electronic microscopy scanning there was one
glomerulus, which revealed expansive mesangium, but no
proliferative mesangial cells or endothelial cells. Part of the
basal membrane demonstrated irregular thickening. There were
magnanimous collagen fibrils (40–60 nm) in the mesangial matrix and
a loose layer of the basal membrane and subendothelial space, with
no electron-dense deposits (Fig. 3).
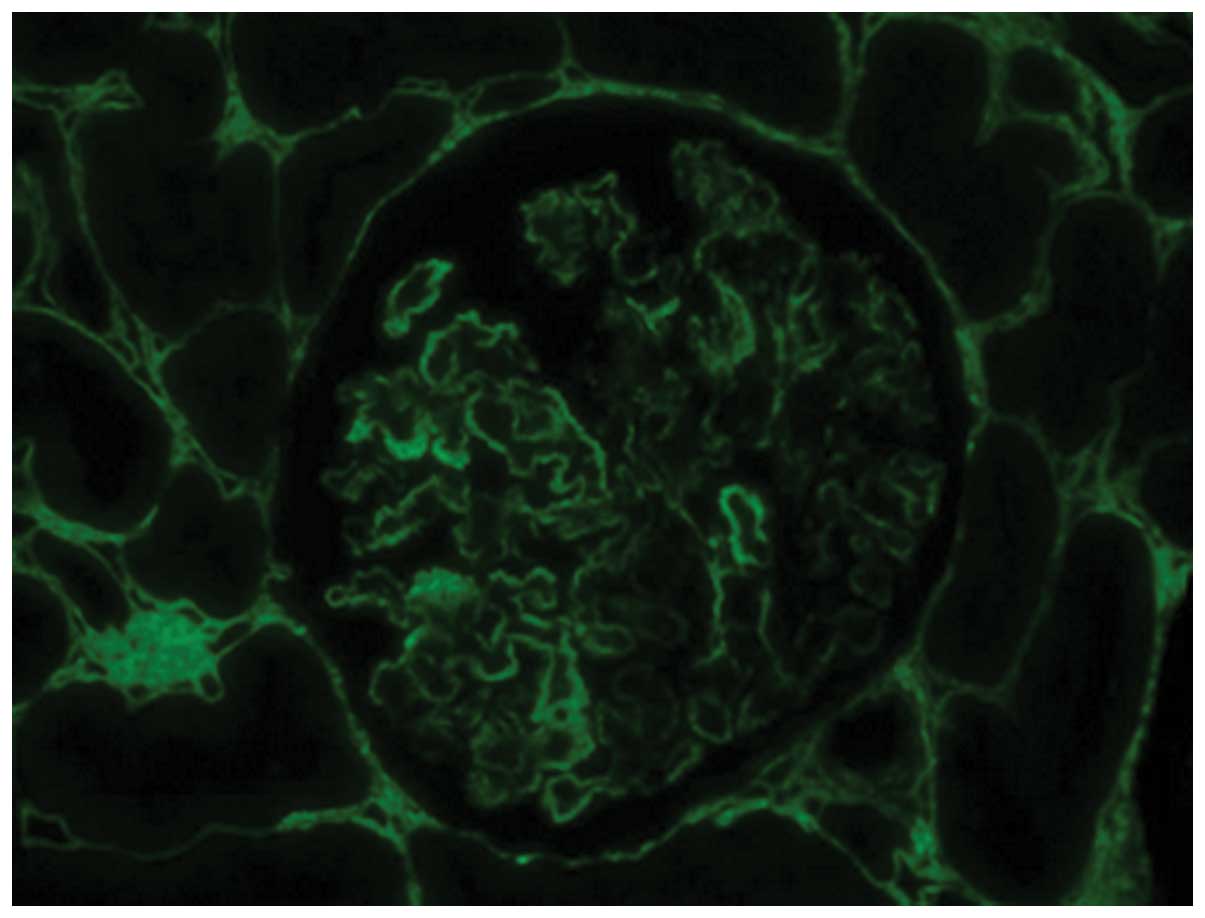
Immunofluorescence staining of the abnormal extracellular infiltrate
for type III collagen was positive (Fig.
4).
Discussion
In the human kidney, type III collagen, a structural
protein of the extracellular matrix, is present only in the
interstitium and blood vessels and not in the glomerulus.
Accumulation of type III collagen fibrils within the mesangial
matrix and subendothelial space is a typical feature of collagen
type III glomerulopathy. No more than forty published cases have
been reported in the English literature (6); the majority are from Japan (2,7–9), with isolated cases from Canada
(10), Italy (3), Slovakia (11) and Brazil (12,13). In
this article, we reported the first case in China with two
completely different renal biopsies.
We described the case of a 12-year-old male, without
a significant family history, who presented with hypertension,
anemia, renal insufficiency and hypocomplementemia. Inherited
factor H deficiency is associated with collagen type III
glomerulopathy and results in hypocomplementemia (14). However, factor H was not detected in
the present case. In the first renal biopsy, there was only segment
and mild mesangial hypercellularity in the glomeruli. No
electron-dense or collagen fibrils were identified by electronic
microscopic scanning. Notably, in the second renal biopsy,
magnanimous collagen fibrils (40–60 nm) in the mesangial matrix and
a loose layer of the basal membrane and subendothelial space were
identified and confirmed as type III collagen fibrils by
immunofluorescence. In the same patient, the results from two renal
biopsies were completely different. We speculate that in the early
stage, type III collagen fibril deposits in the glomeruli were
focal. In the first renal biopsy, only one tiny fragment with four
glomeruli was taken and hence the location where the type III
collagen fibrils were deposited was possibly missed. As the disease
progressed, an increasing number of collagen fibrils were deposited
in the glomeruli. Thus, we were able to diagnose collagen type III
glomerulopathy with the second renal biopsy. Below, we review all
cases in China since 1979 (15–21) for
an improved understanding of collagen type III glomerulopathy.
In China, the cases of 10 female patients and 10
male patients have been reported (Tables
I and II). Three patients had a
family history of kidney disease. The youngest patient was eight
years old and the oldest was 62. The average age of onset of the
disease was 39.50±14.83 years. The early presentation was usually
edema and proteinuria. Seventy-five percent of the patients
developed hypertension. Thirty percent of the patients had
nephrotic syndrome and 20% had mild hematuria. Approximately 50% of
the patients were anemic, and 45% had renal insufficiency. There
was no correlation between creatinine and hemoglobin or blood
pressure. Complement C3 was normal and serum levels of factor H
were not detected in all 20 cases. Notably elevated procollagen
type III peptide was detected in only two cases. The kidneys were
enlarged in the majority of cases.
 | Table I.General information and clinical
manifestations of 20 Chinese cases. |
Table I.
General information and clinical
manifestations of 20 Chinese cases.
| Number | Gender | Age at onset
(years) | Family history | First symptom | Blood pressure
(mmHg) |
|---|
| 1
(20) | Female | 29 | No | Abnormal urine | 140/100 |
| 2
(16) | Male | 33 | Yes | Edema | 175/130 |
| 3
(16) | Male | 34 | Yes | Edema | 190/130 |
| 4
(19) | Male | 55 | No | Edema | 150/85 |
| 5
(17) | Female | 29 | No | Edema | 100/70 |
| 6
(15) | Male | 57 | No | Edema | Normal |
| 7
(15) | Male | 35 | No | Proteinuria | Normal |
| 8
(18) | Male | 62 | No | Proteinuria | 160/100 |
| 9
(21) | Female | 8 | Yes | Edema | 210/130 |
| 10 (21) | Female | 45 | No | Edema | 140/100 |
| 11 (21) | Male | 29 | No | Abnormal urine | 170/105 |
| 12 (21) | Female | 29 | No | Abnormal urine | 140/100 |
| 13 (21) | Female | 47 | No | Abnormal urine | 130/80 |
| 14 (21) | Female | 19 | No | Edema | 140/100 |
| 15 (21) | Male | 39 | No | Abnormal urine | 170/100 |
| 16 (21) | Female | 59 | No | Edema | 160/90 |
| 17 (21) | Female | 56 | No | Abnormal urine | 150/73 |
| 18 (21) | Female | 28 | No | Abnormal urine | 130/80 |
| 19 (21) | Male | 57 | No | Edema,
hypertension | 150/90 |
| 20 (21) | Male | 40 | No | Hypertension | 200/102 |
| Table II.Laboratory results of 20 Chinese
cases. |
Table II.
Laboratory results of 20 Chinese
cases.
| Number | Hematuria
(million/ml) | Proteinuria
(g/l) | Hemoglobin (g/24
h) | Serum creatinine
(µmol/l) | Albumin (g/l) | Cholesterol
(mmol/l) | Triglycerides
(mmol/l) |
|---|
| 1 | 10 |
0.94 | 106 |
71.6 |
37.5 |
5.73 | 2.25 |
| 2 | 0 | 2.1 | 117 | 110 | 38 |
6.65 | 4.17 |
| 3 | 0 |
6.38 | 44 | 313 | 20 |
3.68 | 0.62 |
| 4 | 0 |
24.44 | 134 |
434.3 |
16.8 | 15.1 |
11.83 |
| 5 | 0 |
3.03 | 152 |
Normal | 12 |
12.31 | 2.09 |
| 6 | 0 |
5.6 | 100 | 150 |
23.9 |
4.77 |
2.51 |
| 7 | 0 |
6.5 |
Normal |
Normal | 29 |
4.7 |
1.8 |
| 8 | 0 |
2.6 | 142 | 139 | – | – | – |
| 9 | 0 |
0.24 | 91 |
76.91 |
39.4 |
5.82 |
1.48 |
| 10 | 0 |
0.28 | 129 |
137.91 |
41.4 |
3.13 |
1.01 |
| 11 | 0 |
1.19 | 164 |
89.28 |
50.8 |
5.09 |
2.01 |
| 12 | 0 |
0.77 | 101 |
71.6 |
37.5 |
5.73 |
2.25 |
| 13 | 0 |
1.3 | 118 |
52.16 |
42.7 |
4.38 |
3.68 |
| 14 | 10 |
0.42 | 138 |
53.04 |
35.7 |
5.91 |
1.33 |
| 15 | 0 | 6 | 118 |
119.34 |
33.7 |
6.86 |
2.23 |
| 16 | 0 |
3.27 | 92 |
106.96 |
25.5 |
6.41 |
1.12 |
| 17 | 62 |
3.68 | 91 |
74.26 | 34 |
6.07 |
2.42 |
| 18 | 19 |
1.65 | 105 |
46.85 |
36.3 |
4.58 |
2.49 |
| 19 | 0 |
3.69 | 79 |
174.15 |
27.8 |
7.31 |
2.31 |
| 20 | 30 |
2.13 | 90 |
351.83 |
41.9 |
4.88 |
23.8 |
A renal biopsy was performed in all 20 cases. Under
light microscopy, the volume of the majority of renal glomeruli was
increased without substantial hypercellularity. The glomerular
lesions could be roughly divided into two types; one with nodular
changes without substantial hypercellularity, and the other with no
nodular changes. In both types, the glomerular subendothelial space
appeared loose and widened. Diffuse fake two-track was observed in
the capillary loop. However, in the first type, the subendothelial
space was found to be filled with a homogeneous and eosinophilic
substance by periodic acid-Schiff staining, and filled with a
substance which was dyed green by Masson staining, which caused a
narrowing of the capillary lumina. Foci of interstitial fibrosis
and tubular atrophy were observed in all cases, while arteriolar
hyaline was observed only in cases with hypertension. By
immunofluorescence microscopy, in the majority of cases, antibodies
(IgA, IgG and IgM) and components of the complement (C1q and C3)
were negative. However, IgM or C3 non-specific deposition was
observed in seven cases and IgA was positive in the mesangial area
in four cases. By electronic microscopy scanning, collagen fibrils
(diameter 40–100 nm) were identified in the mesangial matrix and
subendothelial space and type III collagen fibril was confirmed by
immunofluorescence staining. In addition, the fusion of foot
processes was also common. In cases with IgA deposition,
electron-dense material was identified in the mesangial area. No
extra-renal deposition of type III collagen fibrils was identified
in any of the 20 cases.
In early studies, prednisone was used for treatment,
but with little effect (15). In
later studies, the main treatments were angiotensin-converting
enzyme inhibitor and restriction of protein intake. The median
follow-up time in a study of 12 cases was 20.83±7.86 months (6–35
months); new renal insufficiency developed in one patient who had
normal creatinine at diagnosis, while slow progression of renal
dysfunction was observed in four patients who had abnormal
creatinine in the beginning. In one case, malignant hypertension
quickly progressed to end-stage renal disease (21).
In our review, the Chinese patients were mostly
adults, which contrasts with previous reviews where all the
patients are extremely young; six patients were younger than two
years old in a study by Gubler et al (22). Persistent proteinuria is the most
common presentation of collagen type III glomerulopathy. Among
those with proteinuria, 30% of the Chinese patients had nephrotic
syndrome, while 60% of patients fell into the nephritic range in
the English literature (16).
Hypertension was also prominent (in 75% of cases), which is similar
to the results in the English literature (reported in two-thirds of
all cases) (16). Forty-five percent
of patients in China presented with elevated creatinine levels,
while renal function was usually normal or slightly decreased at
presentation in previous studies (6). This may be associated with the late
onset of the disease in Chinese patients.
Complement declining due to a deficiency of factor H
is thought to be a manifestation of collagen type III
glomerulopathy (14), but we noted
that this was rare in Chinese cases. In English publications, cases
have occasionally been described within families (8,22,23);
hence, it has been assumed that collagen type III glomerulopathy
may have an autosomal recessive trait (24). In Chinese literature, cases within
the same family were also noted (16). However, the pathogenesis of collagen
type III glomerulopathy remains unclear. Some consider it to be a
systemic disease (9,24), while others consider it a primary
renal disease (2,3). In China, procollagen type III peptide
increased notably in the cases reported by Chen et al
(16) and cases with coronary heart
disease, thyroid tumor, gallbladder polyps or multiple swollen
lymph nodes in the mediastinum have been reported (21); however, extra-renal type III collagen
fibril deposition was not identified in any of the cases reported.
The pathological manifestation of collagen type III glomerulopathy
is significant (25). The pathology
of collagen type III glomerulopathy in Chinese cases is similar to
that observed in other ethnicities, with the exception that, in
China, certain cases were revealed to be IgA-positive under
immunofluorescence microscopy, and electron-dense material was
identified in the mesangial area. This may be due to the fact that
IgA nephropathy is common in China. It is not hard to establish a
definite diagnosis of collagen type III glomerulopathy with typical
immunohistochemical staining for specific collagen types; however,
there is no effective treatment available.
In conclusion, collagen type III glomerulopathy is a
rare glomerular disease, with 21 cases reported in China to date.
The onset age, clinical manifestation and pathological features of
the disease are not exactly the same worldwide.
References
|
1
|
Arakawa M, Hueki H, Sato M, Yamashita K
and Nakashima S: Idiopathic mesangiodegenerative
glomerulonephropathy: A proposal of a new glomerular disease. Jpn J
Nephrol. 21:14041979.
|
|
2
|
Ikeda K, Yokoyama H, Tomosugi N, Kida H,
Ooshima A and Kobayashi K: Primary glomerular fibrosis: A new
nephropathy caused by diffuse intraglomerular increase in atypical
type III collagen fibers. Clin Nephrol. 33:155–159. 1990.PubMed/NCBI
|
|
3
|
Imbasciati E, Gherardi G, Morozumi K, et
al: Collagen type III glomerulopathy: a new idiopathic glomerular
disease. Am J Nephrol. 11:422–429. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gibson W and More AR: Glomerular
pathology: recent advances. J Pathol. 184:123–129. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Proesmans W, Van Dyck M and Devriendt K:
Nail-patella syndrome, infantile nephrotic syndrome: complete
remission with antiproteinuric treatment. Nephrol Dial Transplant.
24:1335–1338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alchi B, Nishi S and Narita I:
Collagenofibrotic glomerulopathy: clinicopathologic over-view of a
rare glomerular disease. Am J Kidney Dis. 49:499–506. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshioka K, Takemura T, Tohda M, et al:
Glomerular localization of type III collagen in human kidney
disease. Kidney Int. 35:1203–1211. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tamura H, Matsuda A, Kidoguchi N,
Matsumura O, Mitarai T and Isoda K: A family with two sisters with
collagenofibrotic glomerulonephropathy. Am J Kidney Dis. 27:588–595.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yasuda T, Imai H, Nakamoto Y, et al:
Collagenofibrotic glomerulopathy: a systemic disease. Am J Kidney
Dis. 33:123–127. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dombros N and Katz A: Nail-patella like
renal lesion in the absence of skeletal abnormalities. Am J Kidney
Dis. 1:237–240. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bernasovská G, Demes M, Oksa A, et al:
Collagenfibrotic glomerulopathy - rare glomerulonephritis. Vnitr
Lek. 52:1200–1204. 2006.(In Czech). PubMed/NCBI
|
|
12
|
Bichuette Custodio, Castro E, Teixeira V
and Reis MA: Collagenofibrotic glomerulopathy: A description of two
cases. Abstr book World Congress Nephrol. 327:2007.
|
|
13
|
Ferreira RD, Custódio FB, Guimarães CS,
Corrêa RR and Reis MA: Collagenofibrotic glomerulopathy: three case
reports in Brazil. Diagn Pathol. 4:332009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vogt BA, Wyatt RJ, Burke BA, Simonton SC
and Kashtan CE: Inherited factor H deficiency and collagen type III
glomerulopathy. Pediatr Nephrol. 9:11–15. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou FD, Zhou WZ, Huang CX, et al:
Collagen type III glomerulopathy with two case reports. Chin J
Nephrol. 14:75–79. 1998.
|
|
16
|
Chen N, Xu YW, Pan XX, et al: A family
with two brothers with Collagen type III glomerulopathy. Chin J
Nephrol. 21:645–648. 2005.
|
|
17
|
Liu XG, Zhao ZH, Liu Y, Zhang ZG and Guo
MY: Collagen type III glomerulopathy: A case report. Chin J
Nephrol. 21:336–337. 2005.
|
|
18
|
Lu DQ: A case report of Collagen type III
glomerulopathy. Chin J Integr Tradit and Western Nephrol.
6:1132005.
|
|
19
|
Gu FF and Shang HP: Collagen type III
glomerulopathy: A case report. Med J Liaoning. 21:45–46. 2007.
|
|
20
|
Pu QD and Kuang SQ: Collagen type III
glomerulopathy: A case report. China Foreign Med Treat. 23:57–58.
2010.
|
|
21
|
Chen HP, Xu F and Huang Q: Collagen type
III glomerulopathy: clinical and pathological features. J Nephrol
Dialy Transplant. 20:522–529. 2011.
|
|
22
|
Gubler MC, Dommergues JP, Foulard M, et
al: Collagen type III glomerulopathy: a new type of hereditary
nephropathy. Pediatr Nephrol. 7:354–360. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salcedo JR: An autosomal recessive
disorder with glomerular basement membrane abnormalities similar to
those seen in the nail patella syndrome: report of a kindred. Am J
Med Genet. 19:579–584. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soni SS, Gowrishankar S, Nagarik AP, et
al: Collagenofibrotic glomerulopathy in association with Hodgkin's
lymphoma. Saudi J Kidney Dis Transpl. 22:126–129. 2011.PubMed/NCBI
|
|
25
|
Cohen AH: Collagen type III
glomerulopathies. Adv Chronic Kidney Dis. 19:101–106. 2012.
View Article : Google Scholar : PubMed/NCBI
|