Introduction
Laryngeal cancer (LC) is one of the most common
types of head and neck cancer. A large number of protein-coding and
non-coding genes are known to be differentially expressed in LC
cells, indicating a functional switch (1). This gene expression is regulated at the
post-transcriptional level, and is involved in the regulation of
cellular differentiation and development and metabolic processes
(2).
Transcription factors (TFs) and microRNAs (miRNAs)
have crucial roles in the regulation of gene expression (3). TFs are proteins that bind to
cis-regulatory elements located in the upstream regions of genes in
order to activate or suppress transcription, thereby acting to
regulate gene expression at the transcriptional level. TFs may
function alone or in combination with other proteins (4). miRNAs are evolutionarily conserved,
endogenous, non-coding RNAs that post-transcriptionally modulate
gene expression and are associated with tumorigenesis (5). Differentially expressed genes and
miRNAs are significant factors in the development, metastasis and
therapy of LC. miR-21, for example, has been suggested to play an
oncogenic role in the cellular processes of LC (6), and c-Myc is highly expressed at the
protein level in LC (7).
miRNAs target thousands of human genes, known as
target genes; knowledge of these target genes is important for the
analysis of the biological functions of miRNAs (8). A number of databases, including those
that are computationally (9) and
experimentally (10,11) validated, provide an adequate resource
to facilitate an understanding of the associations between miRNAs
and their targets. Certain miRNAs are located within genes known as
host genes, and these miRNAs are transcribed in parallel with their
host transcripts. Two different transcription classes of miRNAs
have been identified: Exonic and intronic (12). Baskerville and Bartel (13) indicated that a close association
exists between intronic miRNA and its host gene. Intronic miRNA and
its host gene are usually expressed in a coordinated manner in
biological progression, and typically work in combination to effect
a biological function and the alteration of signaling pathways
(14). The host gene is considered
in the network of differentially expressed factors when its
corresponding miRNA is differentially expressed. The findings of
the above studies suggest that miRNAs can either work together with
or separately from their host genes in the progression of
cancer.
The aim of the present study was to focus on the
associations among the elements in LC by collecting information on
the differentially expressed and cancer-related genes and miRNAs
involved in LC from databases and literature searches. Three types
of association were considered: miRNAs targeting their targets, TFs
regulating their miRNAs and miRNAs locating within their host
genes. Three networks (differentially expressed, LC-related and
global) were built with the obtained data. Following network
construction, comparisons were made to find the similarities and
differences among the three networks, and the regulatory pathways
involving the differentially expressed elements and common TFs were
separately extracted. We hypothesized that the pathways involving
the differentially expressed elements would have the most
significant influence on the progression of LC, with the abnormal
modulation of these pathways promoting LC development.
Materials and methods
Dataset of experimentally validated
target associations between miRNAs and target genes
The experimentally validated dataset of the gene
targets of each specific miRNA was extracted based on the data
provided by Tarbase 5.0 (10) and
miRTarBase (11). The official
symbols used in this study to unify all miRNAs and genes were
obtained from the National Center for Biotechnology Information
(NCBI) database (http://www.ncbi.nlm.nih.gov/gene/). The inclusion of
these experimental data provides strong evidence to support the
study. The collected results are referred to as dataset
U1.
Dataset of experimentally validated
regulatory associations between TFs and corresponding miRNAs
The dataset of the interaction between the TFs and
miRNAs was collected from TransmiR (15), which is a manually extracted
database. The results from this process are referred to as dataset
U2.
Dataset of miRNAs and host genes
The host genes of respective miRNAs were manually
established based on the data obtained from miRBase (16) and the NCBI. Official symbols and IDs
were used to represent each host gene and miRNA. The collected
results are referred to as dataset U3.
Differentially expressed miRNAs in LC
and LC-related miRNAs
The differentially expressed miRNAs (overexpressed,
downregulated, and upregulated) in LC were mainly extracted from
mir2Disease (17), which is a
manually created database of differentially expressed miRNAs in
various human diseases. In addition, relevant literature was mined
using PubMed (http://www.ncbi.nlm.nih.gov/pubmed) to identify
differentially expressed miRNAs associated with LC, the miRNAs
involved in LC progression, and differentially expressed and
non-differentially expressed miRNAs. The collection of LC-related
miRNAs was completed in the same manner. The collected results are
referred to as dataset U4.
Differentially expressed genes in LC
and LC-related genes
The differentially expressed genes in LC were
gathered from several sources, including Cancer Genetics Web
(http://www.cancerindex.org/geneweb/),
literature searches and the single nucleotide polymorphism (SNP)
database of the NCBI (http://www.ncbi.nlm.nih.gov/snp/). Following the
collection of the differentially expressed gene data, the
LC-related genes were collected from the relevant literature and
the GeneCards database (18). The
P-Match algorithm (19), which
operates on the concepts of pattern matching and weight matrix
approaches to identify TF binding sites (TFBSs) in DNA sequences,
was additionally employed. These TFs were considered to be
LC-related genes and only the TFs that appeared in TransmiR
(15) were focused on. Promoter
region sequences [1,000 nucleotide (nt)] of the targets of
differentially expressed genes were downloaded from the UCSC genome
browser database (20). The P-Match
algorithm was used to identify the TFBSs in the 1,000-nt promoter
region sequences, and these TFBSs were mapped onto the promoter
region of the targets. The matrix library utilized by P-Match
contains sets of known TFBSs collected from the TRANSFAC database,
and thus enables a wide range of TFBSs to be searched for. The
vertebrate matrix and restricted high-quality criterion were used.
The differentially expressed genes that were obtained were included
in the LC-related gene set. The collected results are referred to
as dataset U5.
Network construction at three
levels
Three regulatory networks were constructed to
discuss the regulatory interactions between the genes and miRNAs in
LC. The regulatory associations for the TFs, miRNAs, targets and
host genes from datasets U1, U2
and U3 were extracted. Following the combination
of the associations in these datasets, the global network was
derived. Differentially expressed and LC-related elements were
separately extracted from U4 and
U5, and then separately mapped onto the global
network. The differentially expressed and LC-related networks were
separately derived following the combination of the
associations.
Results
Differentially expressed network in
LC
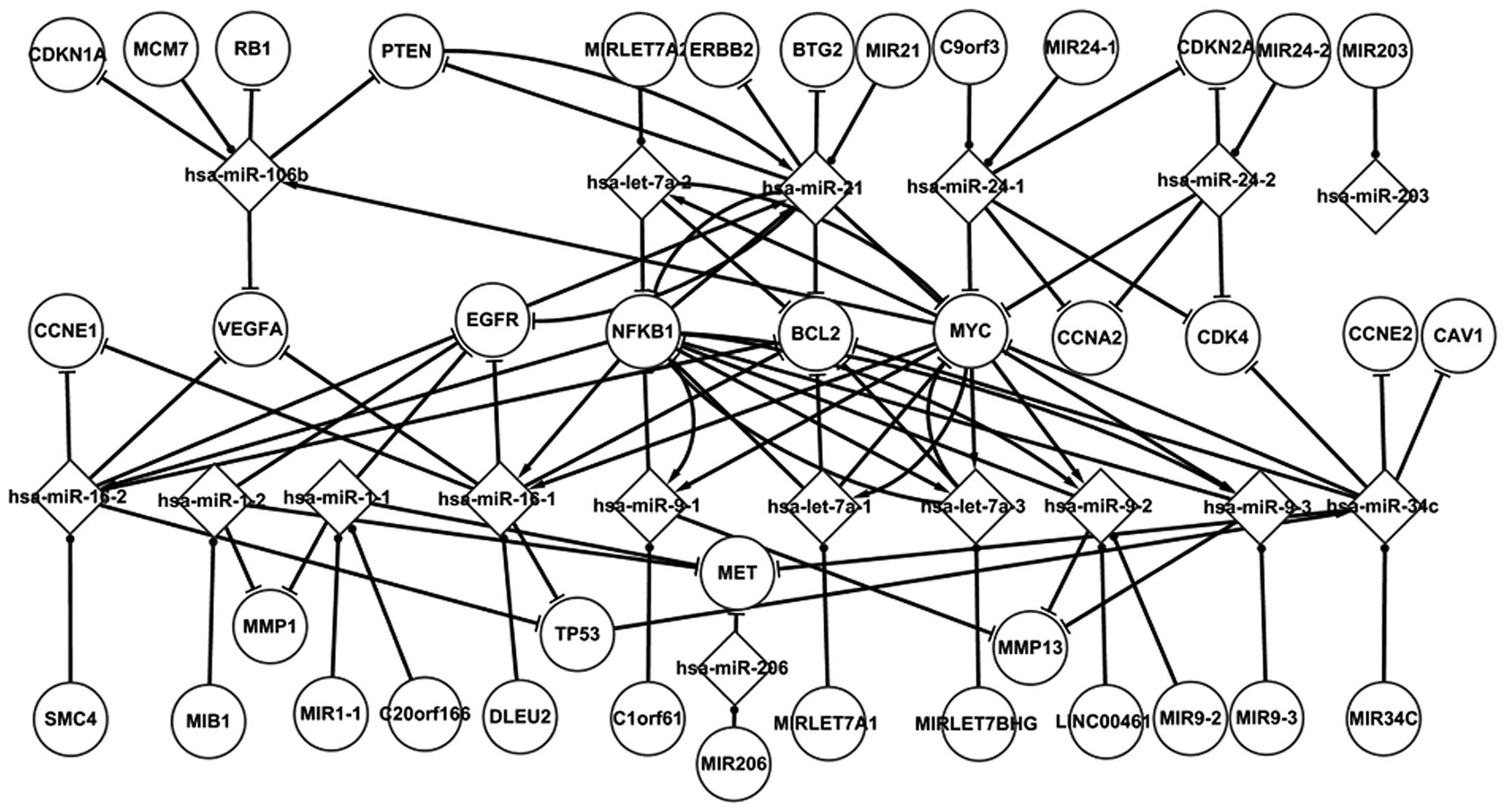
Fig. 1 shows numerous
important regulatory interactions among the differentially
expressed elements in LC. Each node displayed in the figure
represents a differentially expressed gene or miRNA. This network
is composed of five TFs (TP53, PTEN, MYC, EGFR and NFKB1), the
targets of miRNAs, miRNAs and their host genes. With the exception
of the host genes, all the other elements are differentially
expressed in LC. There are three types of associations between the
elements in LC: miRNAs targeting genes, host genes containing
miRNAs and genes regulating miRNAs. Several types of the regulatory
interactions between miRNAs and genes are shown in Fig. 1. Most notable are the five TF-related
pathways. hsa-miR-106b and hsa-miR-21 target PTEN, which regulates
hsa-miR-21. hsa-miR-21 targets EGFR in LC. It is suggested that
PTEN could indirectly influence EGFR through hsa-miR-21. An miRNA
may target one or several genes. hsa-miR-1–2, for example, targets
EGFR, MMP1 and MET. Certain regulatory circuits can also be found
in this network. For example, hsa-let-7a-3 regulates and targets
NFKB1 at the same time. A number of special characteristics of the
host genes and their miRNAs are highlighted in Fig. 1. A host gene may contain one or
several miRNAs, and these miRNAs may target other genes. MIR21, for
example, contains hsa-miR-21, which targets PTEN, MYC, BCL2, EGBB2,
NFKB1, BTG2 and EGFR. The nodes in the differentially expressed
network have already been confirmed by tests. The nodes were
confirmed using numerous sources, including Cancer Genetics Web,
the SNP database of NCBI, and mir2Disease (17). This differentially expressed network
partly reveals the regulatory mechanism of LC.
LC-related network
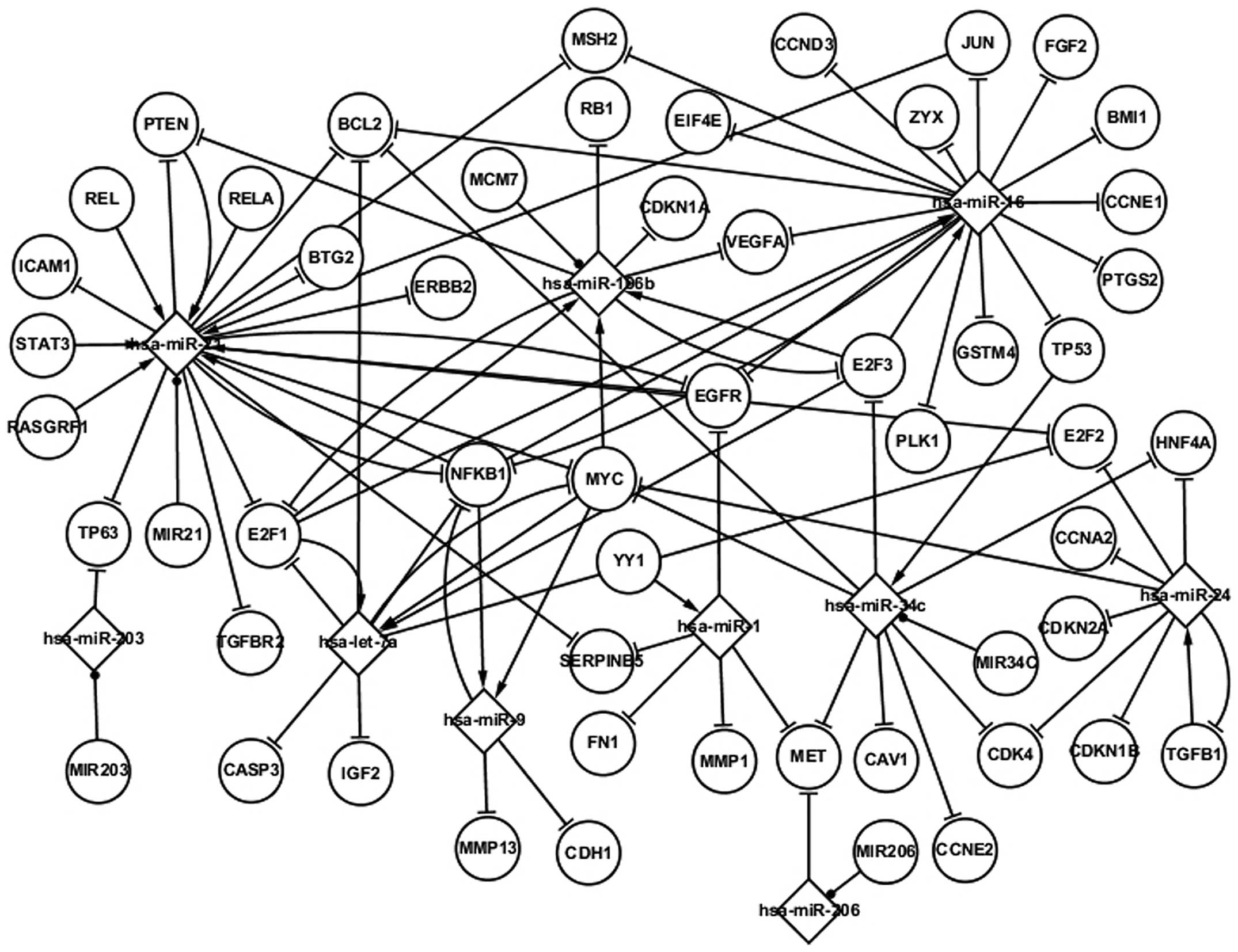
Fig. 2 shows numerous
regulatory interactions among genes and miRNAs in LC. The network
is composed of differentially expressed genes and miRNAs,
LC-related genes and miRNAs, targets of miRNAs and host genes of
miRNAs. The LC-related network can be regarded as an extension of
the differentially expressed network, based on the fact that the
differentially expressed genes and miRNAs are considered to be
LC-related elements. Fig. 2 shows
five differentially expressed TFs (TP53, PTEN, MYC, EGFR and NFKB1)
and nine LC-related TFs (E2F1, E2F3, JUN, RASGRF1, REL, RELA,
STAT3, TGFB1, YY1) in LC. A group of additional pathways can also
be observed in Fig. 2; for example,
STAT3 and RELA regulate hsa-miR-21, which targets ICAM1, and E2F3
and E2F1 regulate hsa-miR-106b, which in turn targets E2F3 and RB1.
YY1 regulates hsa-miR-1, which targets FN1. The LC-related network
shows more topological associations than the differentially
expressed network and contributes to the understanding of the
pathogenesis of LC.
Global network in LC
The global network is the aggregate of datasets
U1, U2 and U3
and includes all the interactions. The global network accommodates
the LC-related network, which additionally contains the
differentially expressed network.
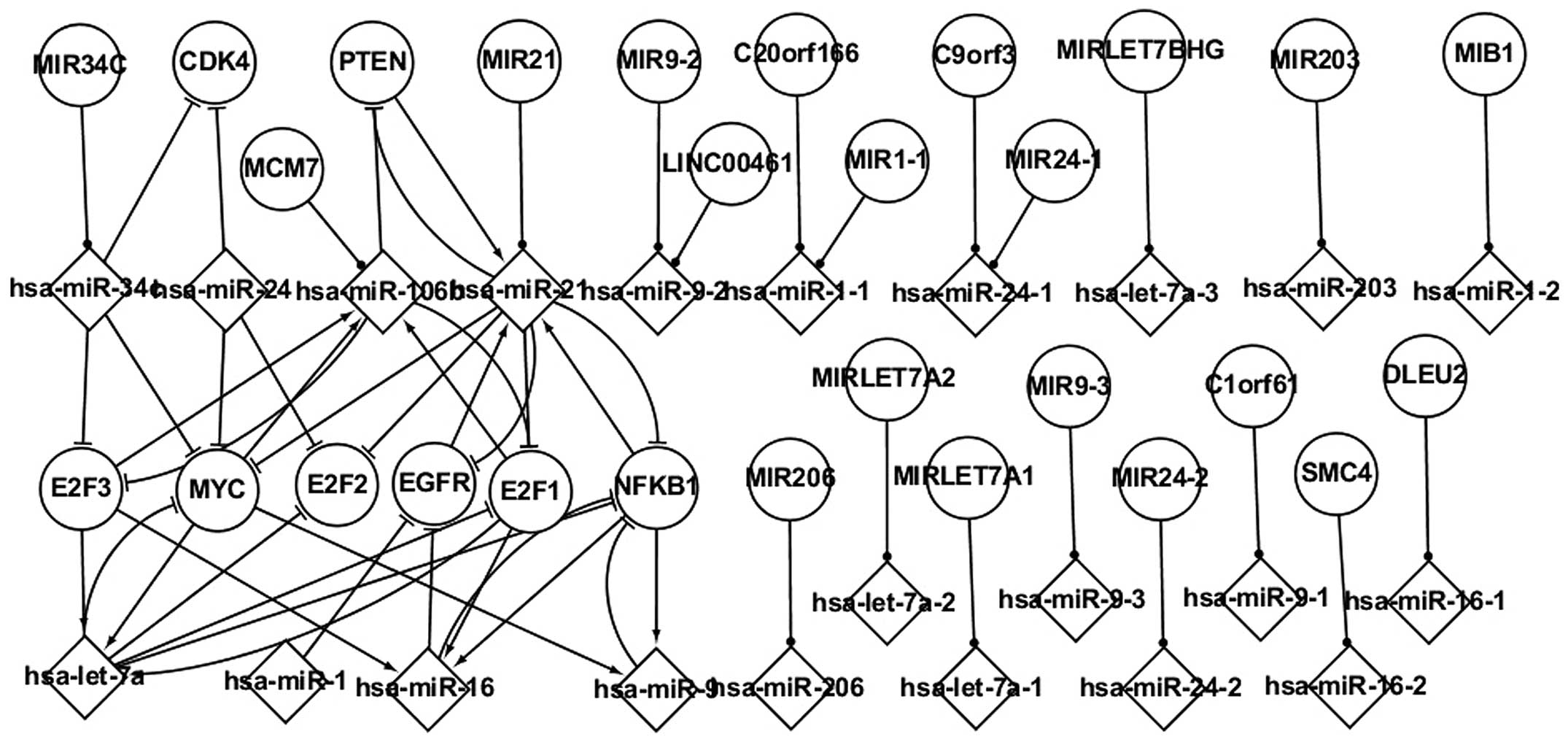
Analysis of host genes and their
miRNAs in LC
Host genes and their miRNAs exhibit certain
important characteristics, and are key biological factors involved
in the regulatory process. Fig. 3
shows a number of pathways involving host genes and their miRNAs.
Although these host genes are not differentially expressed in LC,
their miRNAs are subject to differential expression. Numerous
important regulatory associations exist among the host genes, TFs,
gene targets and the miRNAs contained within the host genes.
MIR34C is the host gene of hsa-miR-34c, which
targets E2F3, MYC and CDK4. MCM7 is the host gene of hsa-miR-106b,
which targets E2F3, E2F1 and PTEN. hsa-miR-106b is regulated by
E2F3, E2F1 and MYC. C1orf61 contains hsa-miR-9-1, which does not
target any gene. hsa-miR-21 is regulated by PTEN, NFKB1 and EGFR,
and hsa-miR-21 and PTEN form a self-adaptation association.
hsa-miR-16 is regulated by E2F3, E2F1 and NFKB1. It is suggested
that an understanding of host genes and their miRNAs could further
aid with the clarification of the pathogenesis underlying LC.
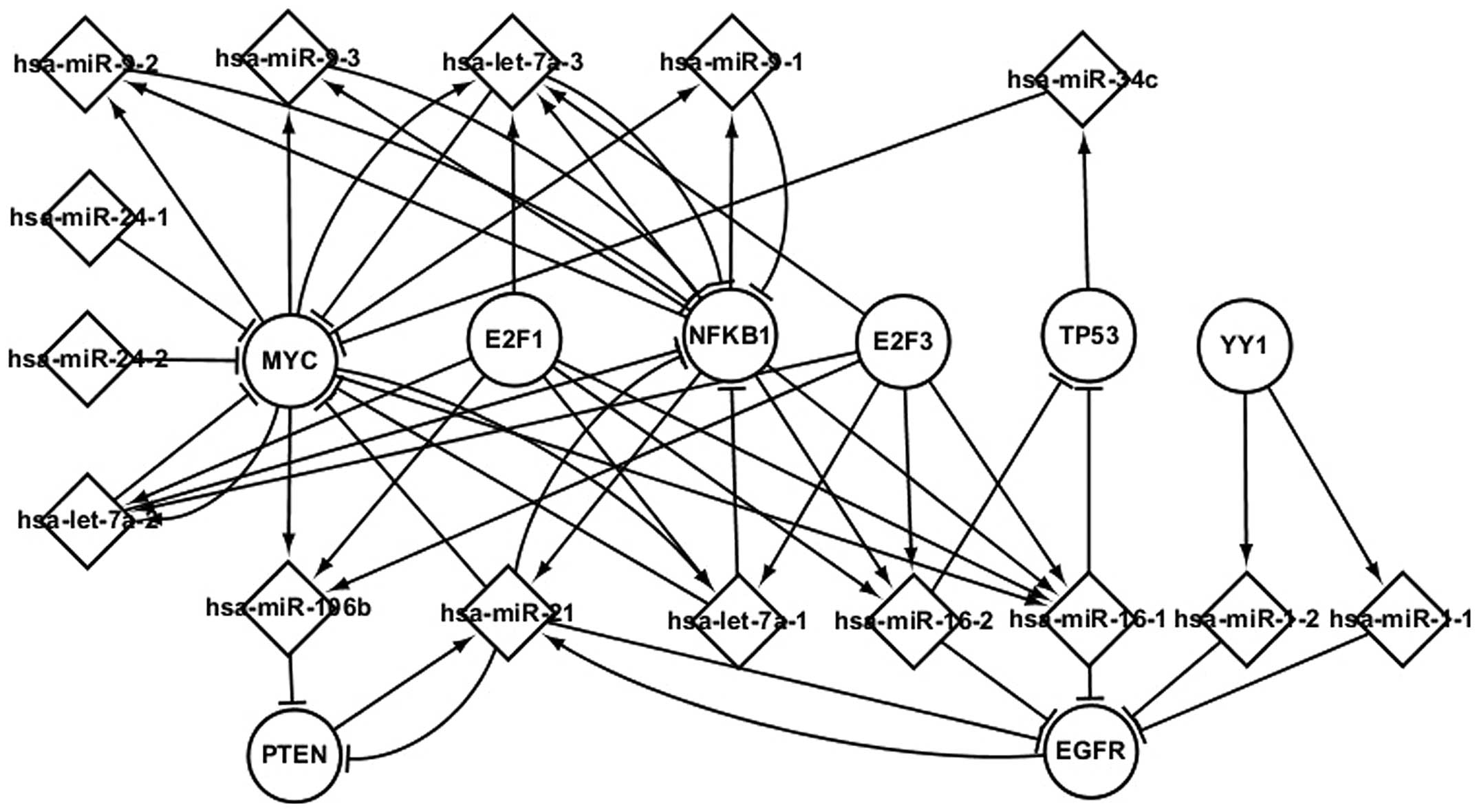
Transcriptional network of TFs and
differentially expressed miRNAs
A further analysis of the differentially expressed
miRNAs exhibiting regulatory interactions with common TFs was
performed. These miRNAs and TFs influence their successors by
targeting and regulating them, respectively. The elements involved
in LC progression are shown in the predicted transcriptional
network in Fig. 4.
Fig. 4 shows the
regulatory interactions among common TFs and miRNAs, as well as
their targets, in LC. The most important TFs are MYC, PTEN, NFKB1
and EGFR. These four TFs and their differentially expressed miRNAs
interact to affect the progression of LC. MYC and NFKB1 coregulate
hsa-let-7a-3 and hsa-9-1, which target NFKB1. As shown in Fig. 4, a differentially expressed miRNA may
target one or several TFs, and a TF may regulate one or several
differentially expressed miRNAs. hsa-miR-16-1, for example, is
regulated by four TFs (E2F3, NFKB1, E2F1 and MYC); five miRNAs
(hsa-miR-21, hsa-miR-16-1, hsa-miR-16-2, hsa-let-7a-1 and
hsa-let-7a-2) target EGFR; and YY1 regulates two miRNAs
(hsa-miR-1-1 and hsa-miR-1–2), which target EGFR. The predicted
transcriptional network showing the TFs and miRNAs is likely to
further enhance the understanding of the pathogenesis of LC.
Comparison and analysis of the
characteristics of the differentially expressed genes in the
networks
In the dataset of the differentially expressed
genes, nodes were classed according to the regulatory association
of adjacent nodes in the three networks in order to compare and
analyze the interaction characteristics of each differentially
expressed gene.
With regard to the TFs, the first class of TF has
six adjacent nodes (three successors and three predecessors). Three
of these TFs, PTEN, TP53 and MYC, exhibit special characteristics,
with both direct predecessors and successors. MYC, for example, is
represented in the three networks, as shown in Table I. MYC is a notable tumor suppressor
with distinct characteristics in the three networks. In the
differentially expressed network, 11 miRNAs target MYC, which
regulates 22 miRNAs; in the LC-related network, 12 miRNAs target
MYC, which regulates 24 miRNAs; and in the global network, 18
miRNAs target MYC, which regulates 32 miRNAs. In the differentially
expressed network, the predecessors indirectly influence the
successors through MYC. As shown in Table I, hsa-miR-17 targets MYC, which in
turn regulates hsa-miR-17, thus generating a self-adaptation
association. This self-adaptation association may be crucial in the
progression of LC. MYC indirectly influences the expression of
other genes via certain miRNAs; for example, MYC regulates
hsa-let-7a-1, which targets BCL2. A number of TFs also indirectly
influence MYC via certain miRNAs; for example TP53 regulates
hsa-miR-34c, which targets MYC. These interactions show that there
are numerous complex associations among MYC, miRNAs and other
genes. In the global network, the involvement of the six previously
mentioned miRNAs (predecessors) and seven additional miRNAs
(successors) in LC has not been validated. Despite this, the
collected information is likely to be of use in the elucidation of
the pathogenesis of LC.
 | Table I.Regulatory interactions between
microRNAs (miRNAs) and MYC in the three networks. |
Table I.
Regulatory interactions between
microRNAs (miRNAs) and MYC in the three networks.
| A, miRNAs targeting
MYC |
|
|
|---|
|
|---|
| Differentially
expressed network | LC-related
network | Global network |
|---|
| hsa-let-7a-1 | hsa-let-7a-1 | hsa-let-7a-1 |
| hsa-let-7a-2 | hsa-let-7a-2 | hsa-let-7a-2 |
| hsa-let-7a-3 | hsa-let-7a-3 | hsa-let-7a-3 |
| hsa-let-7g | hsa-let-7g | hsa-let-7g |
| hsa-miR-145 | hsa-miR-145 | hsa-miR-145 |
| hsa-miR-17 | hsa-miR-17 | hsa-miR-17 |
| hsa-miR-26a-1 | hsa-miR-26a-1 | hsa-miR-26a-1 |
| hsa-miR-34a | hsa-miR-34a | hsa-miR-34a |
| hsa-miR-34b | hsa-miR-34b | hsa-miR-34b |
| hsa-miR-34c | hsa-miR-34c | hsa-miR-34c |
| hsa-miR-24-1 | hsa-miR-21 | hsa-miR-21 |
|
| hsa-miR-24-1 | hsa-miR-24-1 |
|
|
| hsa-miR-20a |
|
|
| hsa-miR-24-2 |
|
|
| hsa-miR-378a |
|
|
| hsa-miR-98 |
|
|
| hsa-miR-26a-2 |
|
|
| hsa-miR-24-2 |
|
|---|
| B, miRNAs regulated
by MYC |
|
|
|
|---|
| Differentially
expressed network | LC-related
network | Global network |
|
|---|
| hsa-let-7a-1 | hsa-let-7a-1 | hsa-let-7a-1 |
| hsa-let-7a-2 | hsa-let-7a-2 | hsa-let-7a-2 |
| hsa-let-7a-3 | hsa-let-7a-3 | hsa-let-7a-3 |
| hsa-let-7b | hsa-let-7b | hsa-let-7b |
| hsa-let-7d | hsa-let-7d | hsa-let-7d |
| hsa-let-7e | hsa-let-7e | hsa-let-7e |
| hsa-let-7g | hsa-let-7g | hsa-let-7g |
| hsa-miR-106 | hsa-miR-106 | hsa-miR-106 |
| hsa-miR-141 | hsa-miR-141 | hsa-miR-141 |
| hsa-miR-15a | hsa-miR-15a | hsa-miR-15a |
| hsa-miR-17 | hsa-miR-17 | hsa-miR-17 |
| hsa-miR-19a | hsa-miR-19a | hsa-miR-19a |
| hsa-miR-221 | hsa-miR-221 | hsa-miR-221 |
| hsa-miR-23b | hsa-miR-23b | hsa-miR-23b |
| hsa-miR-26a-1 | hsa-miR-26a-1 | hsa-miR-26a-1 |
| hsa-miR-29b-1 | hsa-miR-29b-1 | hsa-miR-29b-1 |
| hsa-miR-29b-2 | hsa-miR-29b-2 | hsa-miR-29b-2 |
| hsa-miR-29c | hsa-miR-29c | hsa-miR-29c |
| hsa-miR-34a | hsa-miR-34a | hsa-miR-34a |
| hsa-miR-9-1 | hsa-miR-9-1 | hsa-miR-9-1 |
| hsa-miR-9-2 | hsa-miR-9-2 | hsa-miR-9-2 |
| hsa-miR-9-3 | hsa-miR-9-3 | hsa-miR-9-3 |
|
| hsa-let-7c | hsa-let-7c |
|
| hsa-miR-195 | hsa-miR-195 |
|
|
| hsa-let-7f-1 |
|
|
| hsa-let-7f-2 |
|
|
| hsa-miR-106b |
|
|
| hsa-miR-16-1 |
|
|
| hsa-miR-18a |
|
|
| hsa-miR-19b-1 |
|
|
| hsa-miR-19b-2 |
The second class of TF, which includes EGFR, has
five adjacent nodes (two successors and three predecessors). Six
miRNAs target EGFR, and EGFR regulates hsa-miR-21 in three
networks.
The remaining genes, i.e. those that do not regulate
any miRNAs, can be grouped into one of three classes. In the first
class of gene, each gene has three adjacent nodes (three
predecessors). These genes include CDKN1A, ATM, MMP2 and MMP1,
which are targeted by certain miRNAs but do not regulate any
miRNAs. In the second class of gene, each gene has four types of
adjacent nodes in the three networks (one successor and three
predecessors). These genes include CDKN2A, RB1 and CDK4. In the
third class of gene, each gene again has three adjacent nodes, but
in this case they are three successors. An example of a class-three
gene is FOXM1. It is suggested that the FOXM1 has less of an
impact, as compared with other differentially expressed genes.
Comparison and analysis of the
characteristics of the differentially expressed miRNAs
The first class of miRNA, which includes
hsa-miR-34c, hsa-let-7a-1, hsa-let-7a-2 and hsa-let-7a-3, has six
adjacent nodes (three predecessors and three successors). These
four differentially expressed miRNAs (hsa-miR-34c, hsa-let-7a-1,
hsa-let-7a-2 and hsa-let-7a-3) and their corresponding genes form
four self-adaptation associations. Using hsa-let-7a-1 as an
example, Table II lists the
precursors and successors of the miRNA in the differentially
expressed, LC-related and global networks. In the differentially
expressed network, MYC regulates hsa-let-7a-1, which targets four
differentially expressed genes; in the LC-related network, four
genes regulate hsa-let-7a-1, which targets 13 genes; in the global
network, 10 genes regulate hsa-let-7a-1, which targets 36 genes.
Table II shows that MYC indirectly
influences MYC, CASP8, NRAS and KRAS expression by regulating
hsa-let-7a-1 in the differentially expressed network. MYC and
hsa-let-7a-1 and E2F1 and hsa-let-7a-1 separately form
self-adaptation associations. hsa-let-7a-1 indirectly influences
other miRNAs via certain TFs; for example, hsa-let-7a-1 targets
NFKB1, which regulates hsa-miR-16-1, hsa-miR-16-2, hsa-miR-21 and
hsa-let-7a-3. A number of miRNAs also indirectly influence
hsa-let-7a-1 via certain TFs; for example, hsa-miR-24-1 targets
MYC, which regulates hsa-let-7a-1. These interactions show that
hsa-let-7a-1 has numerous regulatory associations with genes and
other miRNAs.
| Table II.Regulatory interactions between
hsa-let-7a-1 and genes in the three networks. |
Table II.
Regulatory interactions between
hsa-let-7a-1 and genes in the three networks.
| A, Genes regulating
hsa-let-7a-1 |
|
|
|---|
|
|---|
| Differentially
expressed network | LC-related
network | Global network |
|---|
| MYC | MYC | MYC |
|
| E2F1 | E2F1 |
|
| E2F3 | E2F3 |
|
| NFB1 | NFB1 |
|
|
| EIF2C2 |
|
|
| FLI1 |
|
|
| FSH |
|
|
| LIN28 |
|
|
| LIN28B |
|
|
| TRIM32 |
|
| B, Genes targeted
by hsa-let-7a-1 |
|
|
|
| Differentially
expressed network | LC-related
network | Global network |
|
| CASP8 | CASP8 | CASP8, KRAS |
| KRAS | KRAS | MYC, NRAS |
| MYC | MYC | APP, BCL2 |
| NRAS | NRAS | E2F1, E2F2 |
|
| APP | HRAS, IL6 |
|
| BCL2 | ITGB3, NFKB1 |
|
| E2F1 | THBS1, AMMECR1 |
|
| E2F2 | CASP3, CASP9 |
|
| HRAS | CCND2, DICER1 |
|
| IL6 | EGR3, EIF2C4 |
|
| ITGB3 | HMGA1, HMGA2 |
|
| NFKB1 | HNRPDL, IGF2 |
|
| THBS1 | LIN28A, MEIS1 |
|
|
| NEFM, NF2 |
|
|
| NKIRAS2, PRDM1 |
|
|
| RAVER2,
SLC20A1 |
|
|
| TRIM71, TUSC2 |
|
|
| UHRF2, ZFP36L1 |
The second class of miRNA, which includes
hsa-miR-9-1, hsa-miR-9-2 and hsa-miR-9-3, has five adjacent nodes
(two successors and three predecessors). In the differentially
expressed network, MYC and NFKB1 regulate hsa-miR-9, and hsa-miR-9
targets MMP13 and NFKB1; in the LC-related network, MYC and NFKB1
regulate hsa-miR-9, and hsa-miR-9 targets CDH1, MYC and NFKB1.
The third class of miRNA, which includes
hsa-miR-24-1, has four adjacent nodes (three successors and one
predecessor). In the differentially expressed network, hsa-miR-24-1
is not regulated by a TF but targets CDK4, CDKN2A, MYC and CCNA2;
in the LC-related network, TGFB1 regulates hsa-miR-24 and
hsa-miR-24 target seven genes.
The fourth class of miRNA, which includes
hsa-miR-203, has three adjacent nodes (two successors and one
predecessor). In the LC-related network, hsa-miR-203 is not
regulated by a TF but targets TP63.
Comparison and analysis of the
characteristics of common TFs
Following the analysis of the differentially
expressed network, the common TFs in the LC-related network were
compared and analyzed using the same method as that used to compare
the differentially expressed miRNAs. The first class of TF has six
adjacent nodes (three successors and three predecessors). Six TFs
(RUNX1, E2F1, E2F3, NFKB1, YY1 and CREB1) and their corresponding
miRNAs form self-adaptation associations. The following section
focuses on E2F1. Table III shows
the predecessors and successors of E2F1, as well as the associated
regulatory interactions. In the differentially expressed network,
eight miRNAs target E2F1, which in turn regulates eight miRNAs; in
the related network, nine miRNAs target E2F1, which regulates nine
miRNAs; in the global network, 16 miRNAs target E2F1, which
regulates 28 miRNAs. It can be noted from Table III that five miRNAs (hsa-let-7a-1,
hsa-let-7a-2, hsa-let-7a-3, hsa-miR-106a and hsa-miR-17) and E2F1
separately form self-adaptation associations in the three networks.
E2F1 is not differentially expressed in LC, but hsa-let-7a-1,
hsa-let-7a-2 and hsa-let-7a-3 are subject to differential
expression; thus, it can be inferred that the three miRNAs
indirectly lead to changes in the expression of other miRNAs via
E2F1. The pathways involving E2F1 and the differentially expressed
miRNAs indicate that six miRNAs (hsa-miR-149, hsa-miR-23b,
hsa-miR-34a, hsa-miR-15a, hsa-miR-15b and hsa-miR-19a) exhibit
important differential expression in LC. These miRNAs not only are
differentially expressed but also represent adjacent nodes of E2F1
that are frequently involved in transcription in cancer.
| Table III.Regulatory interactions between
microRNAs (miRNAs) and E2F1 in the three networks. |
Table III.
Regulatory interactions between
microRNAs (miRNAs) and E2F1 in the three networks.
| A, miRNAs targeting
E2F1 |
|
|
|---|
|
|---|
| Differentially
expressed network | LC-related
network | Global network |
|---|
| hsa-let-7a-1 | hsa-let-7a-1 | hsa-let-7a-1 |
| hsa-let-7a-2 | hsa-let-7a-2 | hsa-let-7a-2 |
| hsa-let-7a-3 | hsa-let-7a-3 | hsa-let-7a-3 |
| hsa-miR-106a | hsa-miR-106a | hsa-miR-106a |
| hsa-miR-149 | hsa-miR-149 | hsa-miR-149 |
| hsa-miR-17 | hsa-miR-17 | hsa-miR-17 |
| hsa-miR-23b | hsa-miR-23b | hsa-miR-21 |
| hsa-miR-34a | hsa-miR-34a | hsa-miR-23b |
|
| hsa-miR-21 | hsa-miR-34a |
|
|
| hsa-miR-106b |
|
|
| hsa-miR-126 |
|
|
| hsa-miR-20a |
|
|
| hsa-miR-223 |
|
|
| hsa-miR-330 |
|
|
| hsa-miR-93 |
|
|
| hsa-miR-98 |
|
| B, miRNAs regulated
by E2F1 |
|
|
|
| Differentially
expressed network | LC-related
network | Global network |
|
| hsa-let-7a-1 | hsa-let-7a-1 | hsa-let-7a-1 |
| hsa-let-7a-2 | hsa-let-7a-2 | hsa-let-7a-2 |
| hsa-let-7a-3 | hsa-let-7a-3 | hsa-let-7a-3 |
| hsa-miR-106a | hsa-miR-106a | hsa-miR-106a |
| hsa-miR-15a | hsa-miR-15a | hsa-miR-15a |
| hsa-miR-15b | hsa-miR-15b | hsa-miR-15b |
| hsa-miR-17 | hsa-miR-17 | hsa-miR-17 |
| hsa-miR-19a | hsa-miR-19a | hsa-miR-195 |
|
| hsa-miR-195 | hsa-miR-19a |
|
|
| hsa-miR-106b |
|
|
| hsa-let-7i |
|
|
| hsa-miR-16-1 |
|
|
| hsa-miR-16-2 |
|
|
| hsa-miR-18a |
|
|
| hsa-miR-18b |
|
|
| hsa-miR-19b-1 |
|
|
| hsa-miR-19b-2 |
|
|
| hsa-miR-20a |
|
|
| hsa-miR-20b |
|
|
| hsa-miR-223 |
|
|
| hsa-miR-25 |
|
|
| hsa-miR-363 |
|
|
| hsa-miR-449a |
|
|
| hsa-miR-449b |
|
|
| hsa-miR-449c |
|
|
| hsa-miR-92a-1 |
|
|
| hsa-miR-92a-2 |
|
|
| hsa-miR-93 |
The second class of TF, which includes RELA, has
five adjacent nodes (two successors and three predecessors). In the
differentially expressed network, RELA is not targeted by any
differentially expressed miRNAs and it does not regulate any
miRNAs; however, RELA regulates hsa-miR-21 in the LC-related
network.
The third class of TF, which includes STAT1, NKX2–5
and E2F2, has three adjacent nodes (three successors or three
predecessors). In the LC-related network, three differentially
expressed miRNAs target E2F2, which does not, in turn, regulate any
miRNAs.
The fourth class of TF, which includes REL and IRF1,
has two adjacent nodes (two successors and no predecessors). In the
LC-related network, no differentially expressed miRNAs target REL,
but REL regulates hsa-miR-21.
In the final class of TF, which includes TFAP4 and
NF1, each TF has one adjacent node (a predecessor).
Discussion
In the present study, three regulatory topological
networks (differentially expressed, LC-related and global) were
constructed in order to reveal the important pathways in LC and to
establish a topological network regarding the development of LC.
Following construction, the critical hubs and certain notable
features within the networks, including self-adaptation regulation
associations and nodes without direct predecessors or successors,
were taken into consideration. In addition, certain significant
regulatory associations involving differentially expressed genes,
differentially expressed miRNAs and predicted TFs were
identified.
The results of the study demonstrated that four
differentially expressed miRNAs (hsa-let-7a-1, hsa-let-7a-2,
hsa-let-7a-3 and hsa-miR-34c) and their corresponding genes form
self-adaptation associations in LC, while six common TFs (E2F1,
E2F3, NFKB1, RUNX1, YY1 and CRFB1) and their corresponding
differentially expressed miRNAs form self-adaptation associations.
There are numerous pathways involving interactions in which either
one or multiple elements are differentially expressed.
hsa-let-7a-1, for example, targets NFKB1, which in turn regulates
hsa-miR-16-1, and hsa-miR-21 targets MYC, which regulates
hsa-miR-16-1. These pathways perform a key function in LC.
Certain pathways have been found to affect the
progression of LC. It has been suggested that hsa-mir-21 and PTEN
play important roles in the progression of LC, with the two factors
demonstrating a negative correlation (21). Some specific genes, miRNAs, and their
interactions in LC have previously been identified. For example,
the TP53Pro variant may contribute to the risk of LC development
(22). Furthermore, Navarro et
al (23) indicated that TP53
regulates hsa-miR-143 in resected non-small-cell lung cancer. The
present study demonstrated that some pathways are not involved in
LC; however, they influence the progression of numberous other
carcinomas, for example hsa-miR-16 targets BCL2 in chronic
lymphocytic leukemia (24). Although
numerous functions of the identified pathways have not been
elucidated in the present study, the biological functions of these
pathways in other types of cancer may be transferable to LC. The
remaining pathways that have not been detected in cancer may have
some potential functions in LC, for example YY1 regulates
hsa-miR-1-1. For those pathways of predicted TFs, some pathways
have been detected in other carcinomas, for example miR-7 is a
novel inhibitor of YY1 contributing to colorectal tumorigenesis
(25). In this way, information on
the interactions among genes can be extrapolated from one type of
carcinoma to another, and the blind spots of research on the
pathology of LC can be highlighted.
In conclusion, the present study has partly
uncovered the regulatory associations in the development of LC and
supplied comprehensive data associated with LC. These data can be
used to guide medical investigators and biologists undertaking
further research in LC. Investigations into protein interactions
and regulatory patterns (upregulation and downregulation) will
enable the construction of a more comprehensive and extensive
network for LC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 60973091) and the Science
and Technology Development Plan of Jilin Province (no.
20130101166JC).
References
|
1
|
Zhang Y, Chen Y, Yu J, Liu G and Huang Z:
Integrated transcriptome analysis reveals miRNA-mRNA crosstalk in
laryngeal squamous cell carcinoma. Genomics. 104:249–256. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu M, Wu H, Liu T, Li Y, Wang F, Wan H,
Li X and Tang H: Regulation of the cell cycle gene, BTG2, by miR-21
in human laryngeal carcinoma. Cell Res. 19:828–837. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu MH, Xu ZW, Wang KH, Wang N and Li Y:
microRNA and gene networks in human pancreatic cancer. Oncol Lett.
6:1133–1139. 2013.PubMed/NCBI
|
|
5
|
Wang F, Song G, Liu M, Li X and Tang H:
miRNA-1 targets fibronectin1 and suppresses the migration and
invasion of the HEp2 laryngeal squamous carcinoma cell line. FEBS
Lett. 585:3263–3269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren J, Zhu D, Liu M, Sun Y and Tian L:
Downregulation of miR-21 modulates Ras expression to promote
apoptosis and suppress invasion of Laryngeal squamous cell
carcinoma. Eur J Cancer. 46:3409–3416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sang J, Liu L, Tian F, Jin H, Yuan L and
Lou W: The expression of c-myc in the tissues of human laryngeal
squamous cell carcinoma and the effect of siRNA-mediated inhibition
of c-myc on proliferation in laryngeal carcinoma Hep-2 cells. Lin
Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 25:695–700.
2011.PubMed/NCBI
|
|
8
|
Wang K, Xu Z, Wang N, Xu T and Zhu M:
MicroRNA and gene networks in human diffuse large B-cell lymphoma.
Oncol Lett. 8:2225–2232. 2014.PubMed/NCBI
|
|
9
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The microRNA.org resource: Targets and expression.
Nucleic Acids Res. 36(Database): D149–D153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: A functional update of TarBase. Nucleic Acids
Res. 37(Database): D155–D158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu SD, Lin F-M, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39(Database): D163–D169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14(10A): 1902–1910. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRNAs and host genes. RNA. 11:241–247. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cao G, Huang B, Liu Z, Zhang J, Xu H, Xia
W, Li J, Li S, Chen L, Ding H, et al: Intronic miR-301 feedback
regulates its host gene, ska2, in A549 cells by targeting MEOX2 to
affect ERK/CREB pathways. Biochem Biophys Res Commun. 396:978–982.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Lu M, Qiu C and Cui Q: TransmiR: A
transcription factor-micro RNA regulation database. Nucleic Acids
Res. 38(Database): D119–D122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39(Database): D152–D157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37(Database): D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Safran M, Dalah I, Alexander J, Rosen N,
Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al:
GeneCards Version 3: The human gene integrator. Database (Oxford).
2010:baq0202010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chekmenev DS, Haid C and Kel AE: P-Match:
Transcription factor binding site search by combining patterns and
weight matrices. Nucleic Acids Res. 33(Web Server): W432–W437.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujita PA, Rhead B, Zweig AS, Hinrichs AS,
Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A,
et al: The UCSC Genome Browser database: Update 2011. Nucleic Acids
Res. 39(Database): D876–D882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Lei DP, Jin T, Zhao XN, Li G and
Pan XL: Altered expression of miR-21 and PTEN in human laryngeal
and hypopharyngeal squamous cell carcinomas. Asian Pac J Cancer
Prev. 12:2653–2657. 2011.PubMed/NCBI
|
|
22
|
Zemleduch R, Lianeri M, Rydzanicz M,
Gajecka M, Szyfter K and Jagodziński PP: Contribution of
polymorphism in codon 72 of TP53 gene to laryngeal cancer in Polish
patients. Oral Oncol. 45:683–686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Navarro A, Diaz T, Gallardo E, Viñolas N,
Marrades RM, Gel B, Campayo M, Quera A, Bandres E, Garcia-Foncillas
J, et al: Prognostic implications of miR-16 expression levels in
resected non-small-cell lung cancer. J Surg Oncol. 103:411–415.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shumizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang N, Li X, Wu CW, Dong Y, Cai M, Mok
MT, Wang H, Chen J, Ng SS, Chen M, et al: microRNA-7 is a novel
inhibitor of YY1 contributing to colorectal tumorigenesis.
Oncogene. 32:5078–5088. 2013. View Article : Google Scholar : PubMed/NCBI
|