Introduction
Myocardial infarction (MI) is among the leading
causes of mortality worldwide (1).
Patients that survive an acute MI may subsequently suffer heart
failure (HF), with left ventricular remodeling as the most common
underlying factor (2,3). Myocardial hypertrophy, inflammation and
significant fibrosis are considered to be characteristic
pathological alterations of post-infarct remodeling and major
determinants of the progressive deterioration of cardiac function
following MI (4–7).
Generally, remodeling is a complex process modulated
by a cascade of biochemical signaling processes triggered by the
necrotic myocardium (8).
Transforming growth factor-β1 (TGF-β1) is a key inflammatory
molecule that induces cardiac fibrosis (4,9–11). Furthermore, intracellular
mitogen-activated protein kinase (MAPK) signaling cascades have
been demonstrated to serve a crucial function in the pathogenesis
of cardiac hypertrophy and fibrosis, and inhibitors of this
signaling pathway may suppress interstitial fibrosis and improve
cardiac function (12). In addition,
the augmentation of extracellular signal-regulated kinase 1 and 2
(ERK1/2) activity may enhance interstitial fibrosis, myocardial
hypertrophy and cardiac dysfunction (13,14).
Therefore, agents that affect the molecular pathways elicited
during post-infarct remodeling may be promising therapeutic
candidates.
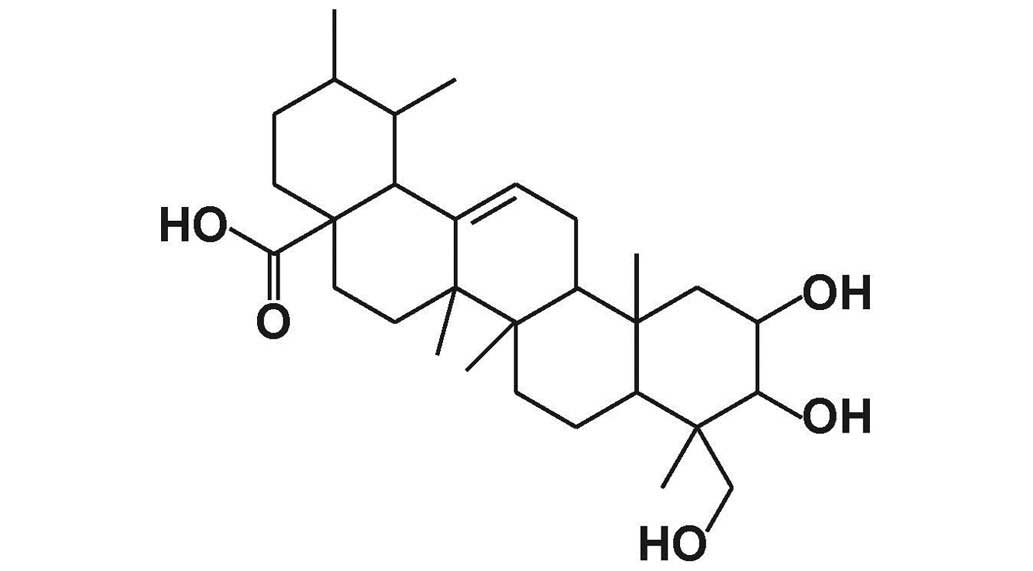
Asiatic acid (AA; Fig.
1) is a pentacyclic triterpenoid derived from the tropical
medicinal plant Centella asiatica (Apiaceae family), and
serves a variety of biological functions (15). AA has been shown to prevent
ultraviolet A-mediated photoaging, facilitate wound healing and
exert anti-inflammatory and anti-diabetic activities (16–19). In
addition, AA has been reported to exhibit anti-fibrotic functions
in the liver via abrogating the TGF-β1/Smad pathway and to protect
the brain from ischemic injury (15,20).
Previous studies have reported that AA exerts beneficial effects on
the heart; for example, AA is able to inhibit pressure
overload-induced cardiac hypertrophy by blocking p38 MAPK and
ERK1/2 phosphorylation and reducing the excessive production of
TGF-β1 and nuclear factor (NF)-κB (21). In addition, AA protects
cardiomyocytes against high-glucose-induced injury by attenuating
oxidative stress (22). However,
whether AA exerts a protective effect on the heart following
myocardial ischemia remains unknown.
Therefore, the present study aimed to investigate
the capacity of AA to inhibit p38 MAPK and ERK1/2 phosphorylation
and TGF-β1 overexpression, thus reducing cardiac inflammation and
fibrosis and improving cardiac function following MI.
Materials and methods
Animals and experimental protocol
All procedures were conducted in accordance with the
Guide for the Care and Use of Laboratory Animals published by the
US National Institutes of Health (8th edition, 2011) and approved
by the Institutional Animal Care and Use Committee of the Academy
of Military Medical Science (SYXK2012-005; Beijing, China). A total
of 120 adult male Sprague-Dawley rats (220–240 g) were purchased
from the Experimental Animal Center, Academy of Military Medical
Science. The rats were maintained in an air-conditioned room at
25±1°C with a 12-h light/dark cycle, and free access to food and
water. After 1 week of acclimatization, rats were allocated at
random into 8 groups (n=15 per group): i) S + V, sham rats treated
with vehicle; ii) S + AA5, sham rats treated with 5
mg/kg/day AA; iii) S + AA25, sham rats treated with 25
mg/kg/day AA; iv) S + AA50, sham rats treated with 50
mg/kg/day AA; v) MI + V, MI rats treated with vehicle; vi) MI +
AA5, MI rats treated with 5 mg/kg/day AA; vii) MI +
AA25, MI rats treated with 25 mg/kg/day AA; and viii) MI
+ AA50, MI rats treated with 50 mg/kg/day AA. MI was
induced by permanent left anterior descending (LAD) coronary artery
ligation. Sham groups underwent an identical surgical procedure
without coronary ligation.
Rat model of MI and AA treatment
Male Sprague-Dawley rats were subjected to left
anterior descending (LAD) coronary artery ligation to induce MI, as
described previously (23). Briefly,
the rats were anaesthetized with 1% sodium phenobarbital (40–60
mg/kg) intraperitoneally and intubated orally with a polyethylene
tube for artificial respiration (UGO Basile S.R.L., Comerio,
Italy). A thoracotomy was performed at the fourth intercostal
space, and the LAD branch was ligated at ~2 mm from its origin.
Sham-operated animals underwent an identical procedure, with the
exception that the coronary ligature was not ligated. Drug
administration was initiated in rats that survived for 6 h after
surgery. AA was dissolved in 5% dimethyl sulfoxide (DMSO) to form a
solution of 10 mg/ml, then diluted to a volume of 2 ml with a
specific concentration determined according to the weight of each
rat. The solution was administered orally to the experimental
groups using an intragastric tube daily for a period of 4 weeks.
The S + V and MI + V groups received an identical volume (2 ml) of
vehicle (5% DMSO) once per day for 4 weeks. To the best of our
knowledge, there are no prior studies associating AA with toxicity
in animal models. However, previous studies have reported that
dosages of 5, 25 or 50 mg/kg/day are well-tolerated by rats and
have no effect on food consumption, activity, weight, mean arterial
pressure and heart rate (24,25).
After 4 weeks of drug administration, the rats were anesthetized,
and lungs were excised and immediately weighed. In addition, the
hearts were excised, washed in ice-cold phosphate-buffered saline
(PBS) and weighed (heart weight, HW).
Echocardiography for determination of
cardiac function
At the end of the 4-week drug administration period,
alterations in left ventricular function were evaluated by
transthoracic echocardiography using a Vivid 7 ultrasound machine
(GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) equipped with a
10-MHz phased-array transducer. Left ventricular systolic diameter
and left ventricular diastolic diameter were measured
simultaneously, and left ventricular ejection fraction (LVEF) and
fractional shortening (FS) were calculated using M-mode recording
by computer algorithms (TruScan 2.07; Harvard Apparatus, Holliston,
MA, USA). All measurements were calculated based on the results of
three consecutive cardiac cycles and all measurements were
performed in a blinded manner (26).
Determination of cardiomyocyte
cross-sectional area
Following 4 weeks of drug administration, the rats
were anesthetized with an intraperitoneal injection of 50 mg/kg
sodium pentobarbital. Adequate anaesthesia was ensured by
monitoring the absence of a withdrawal response to a paw pinch. The
hearts were rapidly excised and washed in ice-cold PBS. Then the
atria were removed and the left ventricle heart samples were fixed
in 4% paraformaldehyde and embedded in paraffin. The heart samples
were cut into 5-µm sections and then stained with hematoxylin and
eosin (H&E; AR1180-100; Wuhan Boster Biological Technology,
Ltd., Wuhan, China). Conventional light microscopy (ML32;
Micro-shot Technology Co., Ltd., Guangzhou, China) at ×400
magnification was used to determine cardiomyocyte cross-sectional
area, as previously described (27).
Determination of tissue cytokines by
enzyme-linked immunosorbent assay (ELISA)
The protein levels of TGF-β1, NF-κB, tumor necrosis
factor (TNF)-α and interleukin (IL)-1β were determined using ELISA
kits (R&D Systems, Inc., Minneapolis, MN, USA), according to
the manufacturer's instructions. In brief, myocardial tissue
samples were homogenized in ice-cold PBS containing protease
inhibitor cocktail (B14001; Biotool LLC, Houston, TX, USA), and
total proteins were extracted using NE-PER Cytoplasmic Extraction
Reagents (Pierce Biotechnology, Inc., Rockford, IL, USA). Total
protein concentration (pg/mg total tissue protein) was determined
using a Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology, Shanghai, China), according to the manufacturer's
instructions.
Determination of infarct size and
cardiac fibrosis
To determine the infarct size of the heart and
interstitial fibrosis in the infarct border zone, the collagenous
fibrotic area of the heart was stained by applying Masson's
Trichrome stain to 5-µm paraffin-embedded sections (28). Infarct size was assessed by examining
images obtained at ×10 magnification using a digital camera (EOS
70D; Canon Co., Ltd., Beijing, China) and calculated as the ratio
of the average scar circumference versus the average inner left
ventricular circumference. The extent of cardiac fibrosis in the
peri-infarct region was assessed by calculating the collagen area
fraction. Quantitative evaluations were performed using ImagePro
Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cardiac myocytes
and cardiac tissues using TRIzol reagent (CW0580A; CWBio Co., Ltd.,
Beijing, China). The cDNA was prepared using a Reverse
Transcription System kit [Promega (Beijing) Biotech Co., Ltd.,
Beijing, China], according to the manufacturer's instructions.
Following reverse transcription, the cDNA obtained was subjected to
qPCR analysis to determine the mRNA expression levels of collagen I
and III. qPCR was performed using predesigned Taqman Gene
Expression Assays and AmpliTaq Gold DNA polymerase kits, following
the manufacturer's instructions (Applied Biosystems, Foster City,
CA, USA). The PCR cycling conditions were as follows: 30 sec at
94°C, 30 sec at 58°C, and 30 sec at 72°C (30 cycles). The mRNA
levels were quantified using SYBR Green (EP1601; BioTeke
Corporation, Beijing, China) and an ABI 7500 Real-Time PCR system
(Applied Biosystems), and GAPDH was used as an internal control.
Primers and probes were designed using Primer Express software,
version 2.0 (Applied Biosystems). Primers used were as follows:
Collagen I, 5′-CAGATTGGGATGGAGGGAGTTTA-3′ (forward) and
5′-CTACAGCACGCTTGTGGATGGCT-3′ (reverse); collagen III,
5′-ATAGCTGAACTGAAAGCCACCAT-3′ (forward) and
5′-CCTGAACTCAAGAGCGGAATA-3′ (reverse); GAPDH,
5′-GTCACCTTCACCGTTCCAGTTTT-3′ (forward) and
5′-CTTAGTTGCGTTACACCCTTTCTT-3′ (reverse).
Western blot assay
Protein was isolated from homogenized heart tissue
following standard protocols (29).
Total proteins were loaded onto a 12% SDS-PAGE gel (P0012A;
Beyotime Institute of Biotechnology) and transferred
electrophoretically to nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). Following blocking with 5% skimmed milk, the
membranes were incubated with the primary antibodies (listed below)
of the recommended dilution at 4°C overnight. The membranes were
washed in Tris-buffered saline and Tween 20 (three times for 10
min) and further incubated with horseradish peroxidase-linked
secondary antibody at 37°C for 60 min. The blots were developed
using an enhanced chemiluminescence reagent kit (EMD Millipore) and
visualized using a Bio-Imaging System (BioSpectrum 510; UVP, LLC,
Upland, CA, USA). VisionWorks LS Acquisition and Analysis Software
were used to analyze blot densities (UVP, LLC).
Primary antibodies used in this study were as
follows: Polyclonal rabbit anti-human ERK1/2 antibody (1:2,000;
sc-292838; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
monoclonal mouse anti-human p-ERK1/2 antibody (1:500; sc-81492;
Santa Cruz Biotechnology, Inc.), monoclonal rabbit anti-human p38
MAPK antibody (1:1,000; #8690; Cell Signaling Technology, Inc.,
Danvers, MA, USA), monoclonal rabbit anti-human phospho-p38 MAPK
antibody (1:1,000; #4511; Cell Signaling Technology, Inc.) and
monoclonal mouse anti-human β-actin antibody (1:5,000; KC-5A08;
KangChen Bio-tech Co., Ltd., Shanghai, China). Secondary antibodies
were polyclonal goat anti-rabbit (1:1,000; HAF008; R&D systems,
Inc.) and polyclonal goat anti-mouse (1:1,000; HAF007; R&D
systems, Inc.).
Statistical analysis
Continuous variables that approximated the normal
distribution were expressed as mean ± standard deviation.
Comparisons between multiple groups were performed using analysis
of variance followed by Bonferroni correction for post hoc t-test.
Two-sided tests were used throughout, and P<0.05 was considered
to indicate a statistically significant difference. SPSS software,
version 14.0 (SPSS, Inc., Chicago, IL, USA) was used for data
analysis.
Results
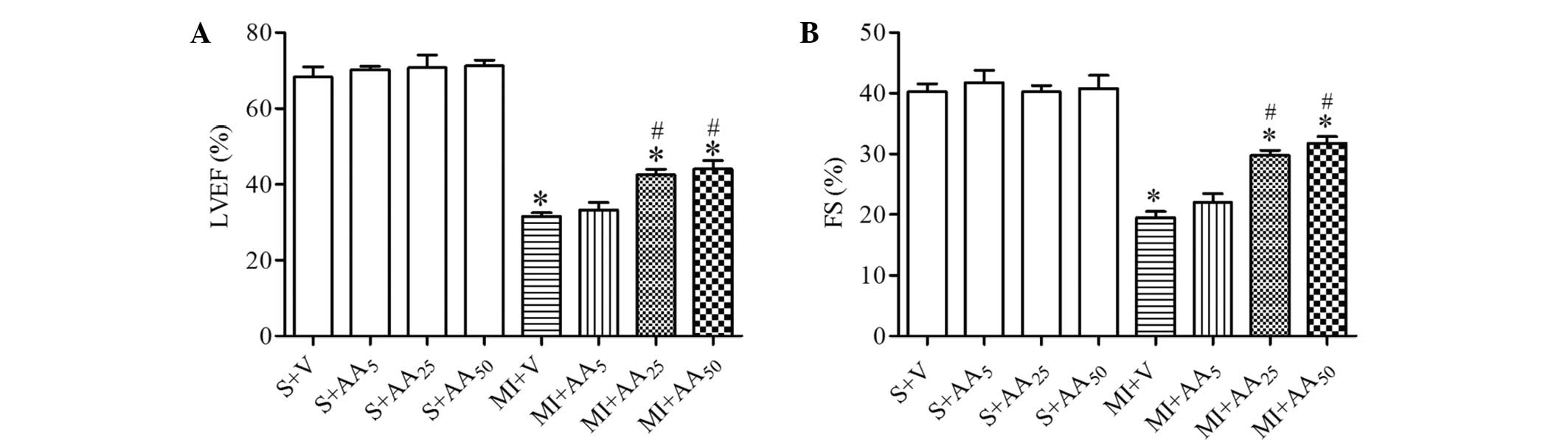
AA (25 mg/kg) exerted a protective
effect on cardiac function after MI
To determine whether AA treatment was able to
improve cardiac function following MI, and to clarify the
appropriate dosage of AA for the further experiments,
echocardiographic analyses were conducted at day 28 post-modeling
(Fig. 2). Compared with the S + V
group, MI injury caused a significant reduction in LVEF (S + V vs.
MI + V, 68.25±2.72 vs. 31.50±1.04%, P<0.05) and FS (S + V vs. MI
+ V, 40.25±1.32 vs. 31.52±1.06%, P<0.05). Treatment with 5 mg/kg
AA produced no significant changes in cardiac function compared
with MI + V rats. However, the MI + AA25 and MI +
AA50 groups exhibited significantly improved cardiac
function compared with the MI + V group, as indicated by increased
LVEF and FS. Additionally, 50 mg/kg AA did not have a greater
cardioprotective effect than 25 mg/kg AA in the MI model rats
(LVEF, MI + AA50 vs. MI + AA25, 44.00±2.27
vs. 42.50±1.44%, P=0.60; FS, MI + AA50 vs. MI +
AA25, 31.75±1.11 vs. 29.53±0.85%, P=0.20). AA exerted a
protective effect on cardiac function following MI in a
dose-dependent manner, and 25 mg/kg AA was selected as the dosage
for the subsequent experiments. None of the three dosages of AA
produced an effect on the echocardiographic parameters in the rats
subjected to sham modeling.
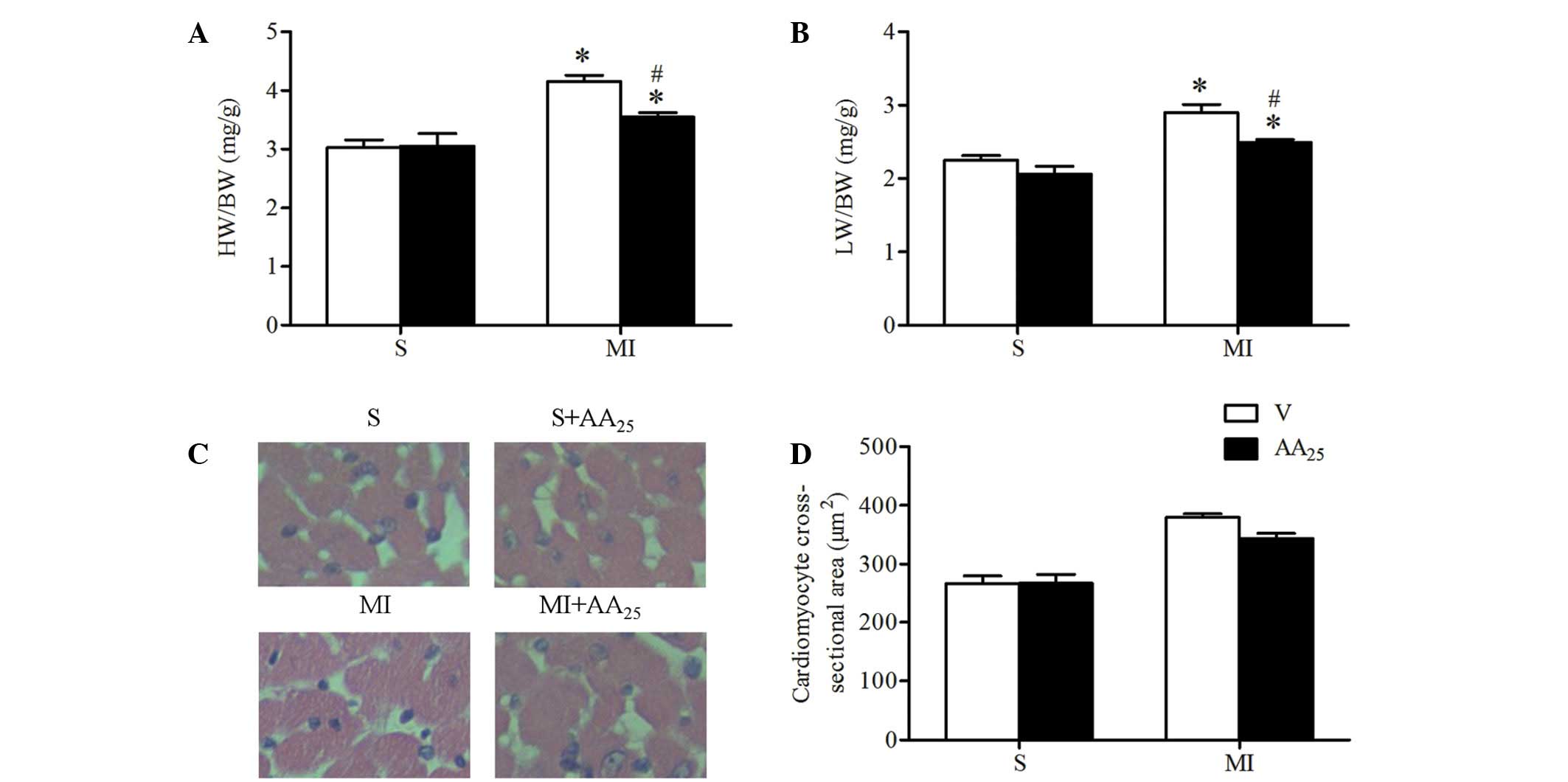
AA prevented cardiac hypertrophy in
response to MI
The HW/body weight ratio of rats subjected to MI was
significantly increased compared with that of the sham rats (MI + V
vs. S + V, 4.16±0.21 vs. 3.03±0.27%; P<0.05), and AA treatment
partly reversed this change (MI + AA25 vs. MI + V,
3.54±0.16 vs. 4.16±0.21%, P<0.05; Fig. 3). The lung wet weight/body weight
ratio of the MI rats was increased compared with that of the sham
rats (MI + V vs. S + V, 2.90±0.22 vs. 2.25±0.13%, P<0.05), and
this change was suppressed by AA treatment (MI + AA25
vs. MI + V, 2.49±0.09 vs. 2.90±0.22%, P<0.05). AA (25 mg/kg) did
not affect cardiac weight in sham rats. These data indicated that
AA prevented cardiac hypertrophy and diminished pulmonary
congestion induced by MI. H&E staining and analysis of the
cardiomyocyte cross-sectional area in myocardial sections of remote
areas to the infarct revealed that MI led to marked cardiomyocyte
hypertrophy (MI + V vs. S + V, 379.3±12.6 vs. 267.0±25.6
µm2, P<0.05), and AA treatment suppressed the
MI-induced cardiomyocyte hypertrophy (MI + AA25 vs. MI +
V: 344.0±17.6 vs. 379.3±12.6 µm2, P<0.05).
AA suppressed the expression of
inflammatory cytokines following MI
The levels of TGF-β1, NF-κB, TNF-α and IL-1β were
tested in left ventricular myocardial tissue lysates using ELISA
kits (Fig. 4). The levels of NF-κB,
TGF-β1, TNF-α and IL-1β were markedly increased in the MI rats
compared with the sham rats, indicating that MI resulted in the
upregulation of proinflammatory cytokine levels. Treatment of the
MI rats with AA markedly reduced the expression levels of TGF-β1,
NF-κB and TNF-α, but had no significant effect on IL-1β expression.
AA produced no effect on the levels of these inflammatory cytokines
in sham rats.
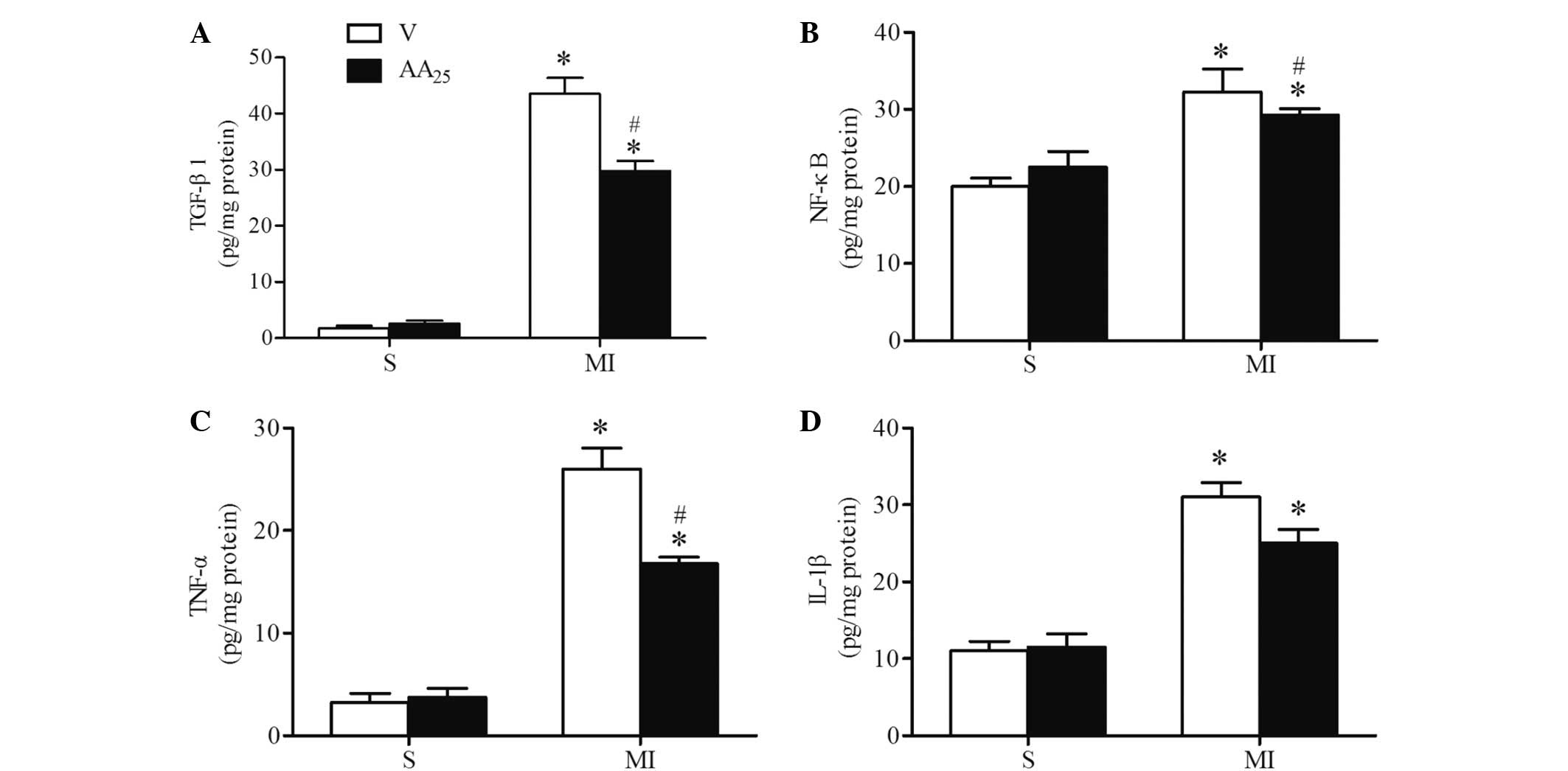 | Figure 4.Expression levels of inflammatory
cytokines in the infarct border zone were suppressed by AA after
MI. Quantitative analysis of the protein expression levels of the
inflammatory cytokines (A) TGF-β1, (B) NF-κB, (C) TNF-α and (D)
IL-1β in four groups (n=6–8 per group) at 4 weeks after MI, as
determined using ELISA. Values are presented as the mean ± standard
deviation. *P<0.05 MI vs. S group; #P<0.05
AA25 vs. V group. V, vehicle; S, sham; TGF-β1,
transforming growth factor-β1; AA, asiatic acid, MI, myocardial
infarction; NF-κB, nuclear factor-κB; TNF-α, tumor necrosis
factor-α; IL-1β, interleukin-1β. ELISA, enzyme-linked immunosorbent
assay |
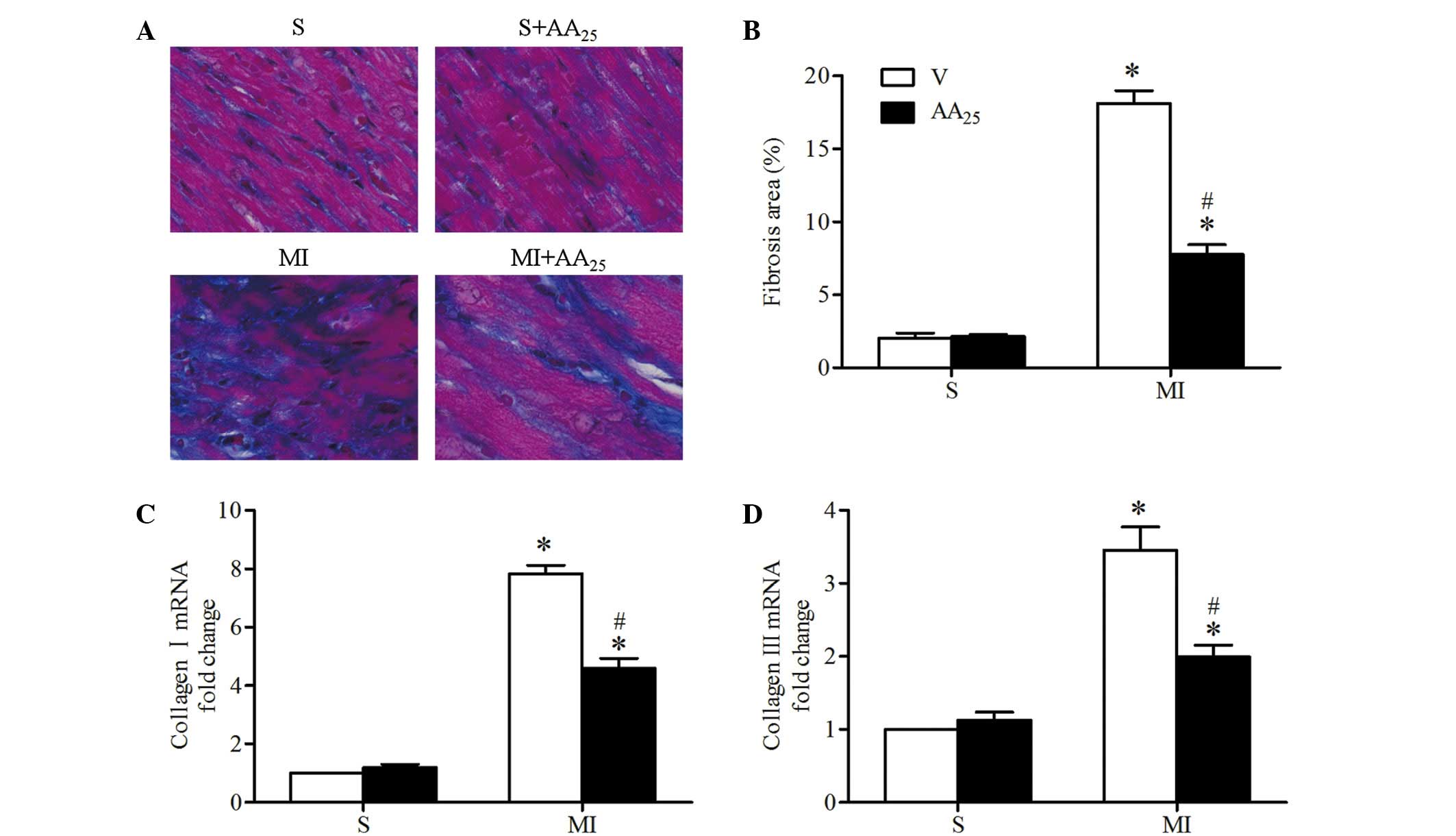
AA treatment prevented post-ischemic
myocardial interstitial fibrosis
Cardiac tissue sections were stained with Masson's
trichrome to determine infarct size of the heart and myocardial
interstitial fibrosis in the infarct border zone (Fig. 5A). Infarct size was comparable
between the vehicle- and AA-treated MI rats, indicating that the
degree of pro-fibrotic stimulation was similar. Mild or no fibrosis
was detected in the sham rats, and MI resulted in significantly
increased interstitial fibrosis (MI + V vs. S + V, 18.13±1.74 vs.
2.03±0.73%, P<0.05; Fig. 5B. AA
treatment markedly decreased the MI-induced fibrosis in the border
zone (MI + AA25 vs. MI + V, 7.78±1.33 vs. 18.13±1.74%,
P<0.05). Consistent with these observations, collagen I and III
mRNA expression levels in the border zone were significantly
increased in MI rats compared with sham rats, and the changes in
the MI rats were reversed by AA treatment (Fig. 5C and D). AA did not affect cardiac
collagen deposits in the sham rats. Collectively, these results
demonstrated that AA prevented interstitial fibrosis in the
myocardial infarct border zone following MI in rats.
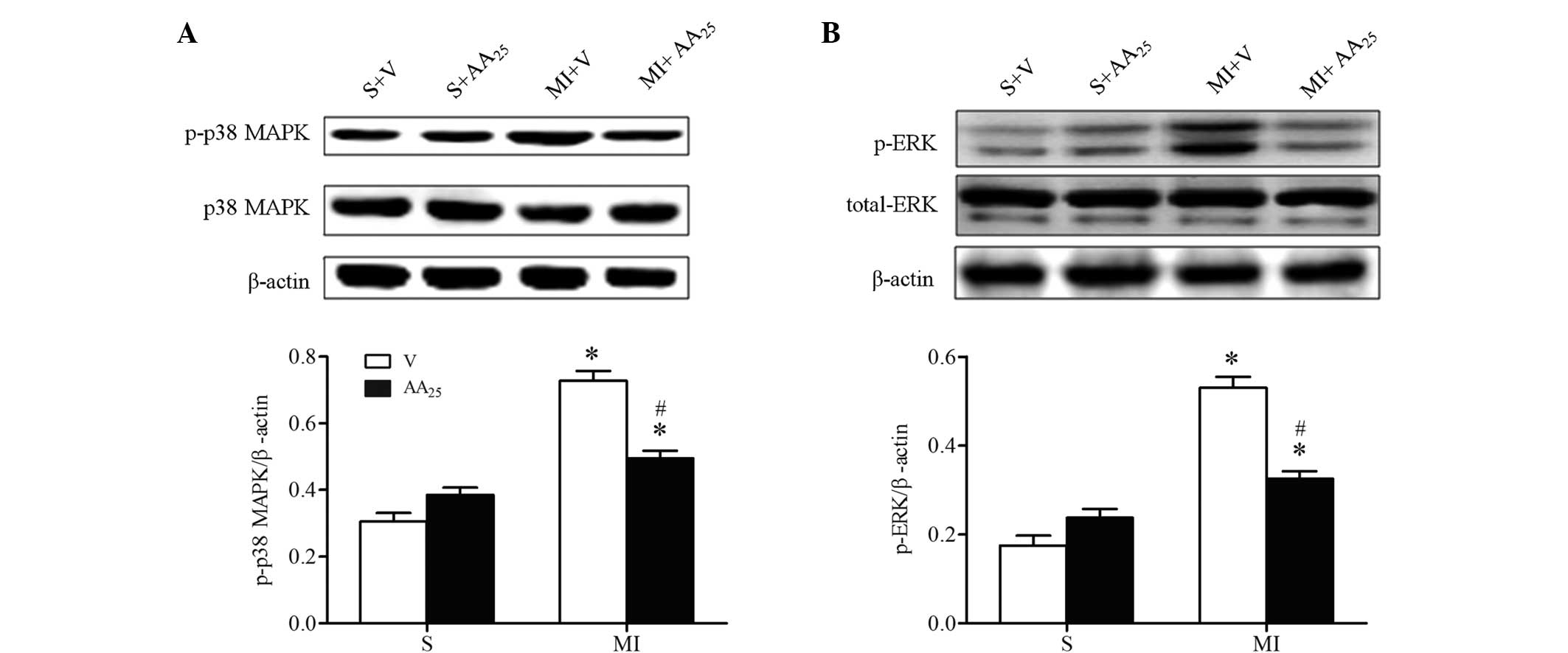
AA inhibited p38 MAPK and ERK1/2
phosphorylation following MI
The aforementioned results suggest that AA
suppressed the expression of inflammatory cytokines in addition to
preventing interstitial fibrosis and myocardial hypertrophy
following MI. To further investigate the molecular mechanisms
underlying the effect of AA on the ischemic heart, the activation
of p38 MAPK and ERK1/2 was determined, as they are reported to
participate in cardiac remodeling (28). As expected, increased phosphorylation
of p38 MAPK and ERK1/2 was observed in the border zone of hearts of
MI rats compared with sham rats, and this change was reversed by AA
treatment (Fig. 6). However, the
total expression levels of p38 MAPK and ERK1/2 were not altered. AA
did not affect the activation of p38 MAPK and ERK1/2 in sham rats.
Collectively, the suppression of the p38 MAPK cascade and the
inhibition of ERK1/2 phosphorylation may account for the beneficial
effects of AA on hearts affected by MI.
Discussion
To the best of our knowledge, the present study is
the first to show that AA treatment preserves cardiac function and
alleviates left ventricular remodeling (30) in rats following MI (31), as indicated by the inhibition of
cardiac hypertrophy, suppression of inflammatory cytokine
expression and prevention of interstitial fibrosis in the infarct
border zone. The inhibition of p38 MAPK and ERK1/2 phosphorylation
may be an underlying mechanism of these effects. The present
results indicate that AA may exert a therapeutic effect against
cardiac dysfunction following MI.
Left ventricular remodeling is a complex process
that occurs following MI, involving cardiomyocyte hypertrophy,
inflammation and fibrosis (4–7).
Initially following MI, fibrosis is necessary to rebuild the
necrotic myocardium to preserve the structural integrity of the
heart and to avoid cardiac rupture. However, the continuous
remodeling of the infarct left ventricle may result in progressive
ventricular dilation, decreased cardiac function and ultimately
heart failure (32,33). The extent of the detrimental
remodeling is a predictive factor for morbidity and mortality
following MI (34). At this stage,
the collagen deposition in the infarct border zone is crucially
involved and contributes substantially to the reduced myocardial
elasticity and impaired cardiac contractility. Furthermore,
hypertrophy and inflammation have been shown to promote myocardial
fibrosis and left ventricular remodeling. Thus, strategies that
inhibit cardiac remodeling following MI represent a major
therapeutic goal of modern cardiology (35).
Recently, herbal compounds such as scutellarin and
Schisandrin B have been demonstrated to produce beneficial effects
against remodeling in the infarcted heart (28,30). The
results of the present study suggest that AA, a compound extracted
from the herb Centella asiatica, may improve impaired
cardiac function following MI. Previous studies have demonstrated
that AA protects cultured cardiomyocytes against high
glucose-induced injury (22) and
exerts anti-hypertrophic effects in a mouse model of cardiac
hypertrophy induced by pressure overload in vivo (21); the mechanisms underlying these
effects may involve the ability of AA to attenuate oxidative stress
and to suppress the protein expression of TGF-β1, NF-κB, p-ERK1/2
and phospho-p38 MAPK. Although these studies clarified the effect
of AA on these signal molecules closely associated with cardiac
remodeling, it remained unclear whether the administration of AA
was able to inhibit cardiac fibrosis and improve cardiac function
following MI. The present study provides the first direct evidence
showing that treatment with AA for 4 weeks significantly inhibits
the deterioration of cardiac function and myocardial remodeling in
a rat model of MI.
The molecular mechanism of left ventricular
remodeling is complex, involving a number of biochemical
intracellular signaling processes. TGF-β1 is an inflammatory
cytokine and a key profibrotic cytokine that is elevated in
experimental MI, and anti-TGF gene therapy attenuates cardiac
remodeling via reducing cardiac fibrosis (31). The present result indicating that AA
inhibited the expression of TGF-β1 suggests that the suppression of
the TGF-β1 pathway underlies the anti-remodeling activity of AA in
post-infarct rat hearts. The p38 MAPK and ERK1/2 pathways, two
classical cascades associated with cardiomyocyte hypertrophy, have
also been demonstrated to serve a crucial function in the
pathogenesis of cardiac remodeling (12–14). Ren
et al demonstrated that daily treatment of rats with the p38
MAPK inhibitor RWJ 67657 resulted in alleviation of cardiac
fibrosis and hypertrophy, in addition to improved cardiac
performance following MI (36). In
addition, Thum et al reported that the activation of ERK1/2
enhanced cardiac fibroblast proliferation and thereby promoted
interstitial fibrosis and cardiac dysfunction (14). The present results revealed that the
activation of p38 MAPK and ERK1/2 induced by MI was significantly
inhibited by the administration of AA, as indicated by the reduced
expression levels of phospho-p38 MAPK and phospho-ERK1/2.
Therefore, the AA-mediated activation of p38 MAPK and ERK1/2
represents a potential mechanism underlying the beneficial effects
of AA on impaired cardiac function following MI.
In conclusion, the results of the present study
demonstrate that AA exerts notable benefits on impaired cardiac
function and heart remodeling following MI, and that the
suppression of TGF-β1, phospho-p38 MAPK and phospho-ERK1/2
expression may underlie these effects. These findings suggest that
AA therapy may be effective in the treatment of ischemia-associated
heart diseases.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 2012BAJ18B01). The authors
thank the Academy of Military Medical Science for their
assistance.
References
|
1
|
Roger VL, Go AS, Lloyd-Jones DM, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
Heart disease and stroke statistics-2012 update: A report from the
American Heart Association. Circulation. 125:e2–e220. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Opie LH, Commerford PJ, Gersh BJ and
Pfeffer MA: Controversies in ventricular remodelling. Lancet.
367:356–367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Biomarkers, genomics, telemetry,
computational biology and zebrafish: Will one of these solve the
problems of post-myocardial infarction heart failure? Eur Heart J.
35:57–58. 2014.PubMed/NCBI
|
|
4
|
Khan R and Sheppard R: Fibrosis in heart
disease: Understanding the role of transforming growth factor-beta
in cardiomyopathy, valvular disease and arrhythmia. Immunology.
118:10–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mann DL: Mechanisms and models in heart
failure: A combinatorial approach. Circulation. 100:999–1008. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fosshaug LE, Berge RK, Beitnes JO, Berge
K, Vik H, Aukrust P, Gullestad L, Vinge LE and Øie E: Krill oil
attenuates left ventricular dilatation after myocardial infarction
in rats. Lipids Health Dis. 10:2452011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang F, Yang XP, Liu YH, Xu J, Cingolani
O, Rhaleb NE and Carretero OA: Ac-SDKP reverses inflammation and
fibrosis in rats with heart failure after myocardial infarction.
Hypertension. 43:229–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jugdutt BI: Aging and remodeling during
healing of the wounded heart: Current therapies and novel drug
targets. Curr Drug Targets. 9:325–344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nicoletti A, Heudes D, Mandet C, Hinglais
N, Bariety J and Michel JB: Inflammatory cells and myocardial
fibrosis: Spatial and temporal distribution in renovascular
hypertensive rats. Cardiovasc Res. 32:1096–1107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H, Wu J, Dong H, Khan SA, Chu ML and
Tsuda T: Fibulin-2 deficiency attenuates angiotensin II-induced
cardiac hypertrophy by reducing transforming growth factor-β
signalling. Clin Sci (Lond). 126:275–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Muslin AJ: MAPK signalling in
cardiovascular health and disease: Molecular mechanisms and
therapeutic targets. Clin Sci (Lond). 115:203–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yeh CC, Li H, Malhotra D, Turcato S,
Nicholas S, Tu R, Zhu BQ, Cha J, Swigart PM, Myagmar BE, et al:
Distinctive ERK and p38 signaling in remote and infarcted
myocardium during post-MI remodeling in the mouse. J Cell Biochem.
109:1185–1191. 2010.PubMed/NCBI
|
|
14
|
Thum T, Gross C, Fiedler J, Fischer T,
Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et
al: MicroRNA-21 contributes to myocardial disease by stimulating
MAP kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krishnamurthy RG, Senut MC, Zemke D, Min
J, Frenkel MB, Greenberg EJ, Yu SW, Ahn N, Goudreau J, Kassab M, et
al: Asiatic acid, a pentacyclic triterpene from Centella
asiatica, is neuroprotective in a mouse model of focal cerebral
ischemia. J Neurosci Res. 87:2541–2550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang SS, Chiu CS, Chen HJ, Hou WC, Sheu
MJ, Lin YC, Shie PH and Huang GJ: Antinociceptive activities and
the mechanisms of anti-inflammation of asiatic acid in mice. Evid
Based Complement Alternat Med. 2011:8958572011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramachandran V, Saravanan R and
Senthilraja P: Antidiabetic and antihyperlipidemic activity of
asiatic acid in diabetic rats, role of HMG CoA: In vivo and in
silico approaches. Phytomedicine. 21:225–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bian D, Zhang J, Wu X, Dou Y, Yang Y, Tan
Q, Xia Y, Gong Z and Dai Y: Asiatic acid isolated from Centella
asiatica inhibits TGF-β1-induced collagen expression in human
keloid fibroblasts via PPAR-γ activation. Int J Biol Sci.
9:1032–1042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong MS, Jung SH, Kim HJ, Kim JR, Zhao LX,
Lee ES, Lee EJ, Yi JB, Lee N, Cho YB, et al: Structure-related
cytotoxicity and anti-hepatofibric effect of asiatic acid
derivatives in rat hepatic stellate cell-line, HSC-T6. Arch Pharm
Res. 27:512–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Si L, Xu J, Yi C, Xu X, Wang F, Gu W,
Zhang Y and Wang X: Asiatic acid attenuates cardiac hypertrophy by
blocking transforming growth factor-β1-mediated hypertrophic
signaling in vitro and in vivo. Int J Mol Med. 34:499–506.
2014.PubMed/NCBI
|
|
22
|
Chan CY, Mong MC, Liu WH, Huang CY and Yin
MC: Three pentacyclic triterpenes protect H9c2 cardiomyoblast cells
against high-glucose-induced injury. Free Radic Res. 48:402–411.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Orenstein TL, Parker TG, Butany JW,
Goodman JM, Dawood F, Wen WH, Wee L, Martino T, McLaughlin PR and
Liu PP: Favorable left ventricular remodeling following large
myocardial infarction by exercise training. Effect on ventricular
morphology and gene expression. J Clin Invest. 96:858–866. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bunbupha S, Pakdeechote P, Kukongviriyapan
U, Prachaney P and Kukongviriyapan V: Asiatic acid reduces blood
pressure by enhancing nitric oxide bioavailability with modulation
of eNOS and p47phox expression in L-NAME-induced hypertensive rats.
Phytother Res. 28:1506–1512. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee KY, Bae ON, Serfozo K, Hejabian S,
Moussa A, Reeves M, Rumbeiha W, Fitzgerald SD, Stein G, Baek SH, et
al: Asiatic acid attenuates infarct volume, mitochondrial
dysfunction, and matrix metalloproteinase-9 induction after focal
cerebral ischemia. Stroke. 43:1632–1638. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Araki S, Izumiya Y, Hanatani S, Rokutanda
T, Usuku H, Akasaki Y, Takeo T, Nakagata N, Walsh K and Ogawa H:
Akt1-mediated skeletal muscle growth attenuates cardiac dysfunction
and remodeling after experimental myocardial infarction. Circ Heart
Fail. 5:116–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vahtola E, Louhelainen M, Merasto S,
Martonen E, Penttinen S, Aahos I, Kytö V, Virtanen I and Mervaala
E: Forkhead class O transcription factor 3a activation and Sirtuin1
overexpression in the hypertrophied myocardium of the diabetic
Goto-Kakizaki rat. J Hypertens. 26:334–344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan Z, Zhao W, Zhang X, Wang B, Wang J,
Sun X, Liu X, Feng S, Yang B and Lu Y: Scutellarin alleviates
interstitial fibrosis and cardiac dysfunction of infarct rats by
inhibiting TGFβ1 expression and activation of p38-MAPK and ERK1/2.
Br J Pharmacol. 162:688–700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu D, Wang H, Zhang J, Zhang X, Xin C,
Zhang F, Lee Y, Zhang L, Lian K, Yan W, et al: Irisin improves
endothelial function in type 2 diabetes through reducing
oxidative/nitrative stresses. J Mol Cell Cardiol. 87:138–147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen P, Pang S, Yang N, Meng H, Liu J,
Zhou N, Zhang M, Xu Z, Gao W, Chen B, et al: Beneficial effects of
schisandrin B on the cardiac function in mice model of myocardial
infarction. PLoS One. 8:e794182013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okada H, Takemura G, Kosai K, Li Y,
Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, et
al: Postinfarction gene therapy against transforming growth
factor-beta signal modulates infarct tissue dynamics and attenuates
left ventricular remodeling and heart failure. Circulation.
111:2430–2437. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kurrelmeyer K, Kalra D, Bozkurt B, Wang F,
Dibbs Z, Seta Y, Baumgarten G, Engle D, Sivasubramanian N and Mann
DL: Cardiac remodeling as a consequence and cause of progressive
heart failure. Clin Cardiol. 21:I14–I19. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hein S, Arnon E, Kostin S, Schönburg M,
Elsässer A, Polyakova V, Bauer EP, Klövekorn WP and Schaper J:
Progression from compensated hypertrophy to failure in the
pressure-overloaded human heart: Structural deterioration and
compensatory mechanisms. Circulation. 107:984–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
White HD and Braunwald E: Applying the
open artery theory: Use of predictive survival markers. Eur Heart
J. 19:1132–1139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vanhoutte D, Schellings M, Pinto Y and
Heymans S: Relevance of matrix metalloproteinases and their
inhibitors after myocardial infarction: A temporal and spatial
window. Cardiovasc Res. 69:604–613. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ren J, Zhang S, Kovacs A, Wang Y and
Muslin AJ: Role of p38alpha MAPK in cardiac apoptosis and
remodeling after myocardial infarction. J Mol Cell Cardiol.
38:617–623. 2005. View Article : Google Scholar : PubMed/NCBI
|