Introduction
Myocardial reperfusion is a therapeutic strategy
which is commonly used to treat patients with acute coronary
syndrome; however, reperfusion following ischemia may promote
additional cellular injury, which accounts for ~50% of the final
size of a myocardial infarct (1). A
previous study demonstrated that ischemia/reperfusion (I/R) may
promote the production of numerous proinflammatory cytokines,
neutrophil infiltration and the apoptosis of cardiomyocytes, which
is likely to result in cardiomyocyte damage (1). Interleukin (IL)-17A, which is a member
of the IL-17 cytokine family, is secreted by CD4+ αβ T
cells, rδ T cells, natural killer cells, and neutrophils (2). Previous studies have demonstrated that
IL-17A may stimulate the production of numerous proinflammatory
mediators, including cytokines, chemokines and matrix
metalloproteinases (3–5). It has been demonstrated that IL-17A is
primarily produced by rδ T cells following myocardial I/R injury,
and IL-17A has a pathogenic role by inducing neutrophil
infiltration and the apoptosis of cardiomyocytes (6). Therefore, IL-17A may facilitate the
release of proinflammatory cytokines and thus may have a key role
in myocardial I/R injury.
Previous studies have demonstrated that vagal
stimulation (VS) and acetylcholine (ACh) are capable of exerting
cardioprotective effects in various cardiovascular diseases
(7,8). Furthermore, the inhibition of
inflammation has been demonstrated to be one of the major
pathophysiological mechanisms by which VS induces cardioprotection
(9,10). As a proinflammatory cytokine, IL-17A
interacts with other proinflammatory mediators, including tumor
necrosis factor (TNF)-α, IL-1β and IL-6 (11); therefore, VS and IL-17A may be
associated with the regulation of apoptosis in cardiomyocytes
(6,12). The authors of the present study
hypothesized that there may be a cardioprotective association
between IL-17A and VS. In the present study, the hypothesis that VS
may attenuate myocardial I/R injury by suppressing IL-17A
expression levels was investigated in a rat model of myocardial
I/R.
Materials and methods
Animal preparation and experimental
design
The present study conformed to the Guideline for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH Publication, revised 1996) (13) and was approved by the Renmin Hospital
of Wuhan University Animal Care and Use Committee (Wuhan, China). A
total of 48 male Sprague-Dawley rats, weighing 250–300 g, were
supplied by the Experimental Animal Center of Vital River
Laboratories (Beijing, China). The rats were randomized into four
equal groups, as follows: Group 1, sham operated (SO) rats were
subjected to surgical manipulation without the induction of
myocardial ischemia (n=12); group 2, I/R rats were subjected to
occlusion of the left anterior descending coronary artery (LAD) for
30 min, followed by reperfusion for 4 h (n=12); group 3, I/R + VS
(n=12) rats were treated the same as group 2, with the exception
that VS was initiated 15 min post-ischemia and lasted for 30 min
(9); and group 4, I/R + anti-IL-17A
(n=12) rats were treated the same as group 2, with the exception
that 200 µg anti-IL-17A neutralized monoclonal antibodies (mAbs)
(Biosynthesis Biotechnology Co., Ltd., Beijing, China) were
intravenously injected 30 min prior to occlusion of the LAD
(4).
The rats were anesthetized with sodium pentobarbital
(45 mg/kg, i.p.), fixed on an electric heating pad to maintain
their body temperature at 37°C, and subsequently artificially
ventilated with a volume-controlled rodent respirator at 70
strokes/minute. Lead II of the electrocardiogram was recorded using
subcutaneous stainless steel electrodes. The electrocardiogram was
monitored during operation and vagal stimulation using a biological
signal recording system (MP150; BIOPAC Systems, Inc., Goleta, CA,
USA)
In the SO, I/R and anti-IL-17A groups, rats
underwent right cervical vagus nerve trunk exposure without
stimulation. In the VS group, the right cervical vagus nerve trunk
was isolated from the surrounding tissue with a small piece of
parafilm. A pair of bipolar platinum electrodes, covered with
warmed paraffin oil for insulation, was placed on the vagus nerve
trunk and the vagal nerve was subsequently stimulated by
rectangular electrical pulses at 10 Hz for 2 msec using a S88 Nerve
and Muscle Stimulator (Grass Technologies; Natus Neurology Inc.,
Warwick, RI, USA). The electrical voltages of the pulses were
optimized in each rat during stimulation in order to obtain a 10%
reduction in heart rate from the baseline values. The actual
stimulus voltage was 1–7 V.
The rats underwent a 30-min LAD occlusion, followed
by reperfusion for 4 h (14).
Briefly, a left thoracotomy and pericardiectomy were performed in
order to expose the anterior wall of the left ventricle. Using a
small curved needle, a 5-0 silk suture was subsequently passed
through the myocardium beneath the middle segment of the LAD branch
coursing down the middle of the anterior wall of the left
ventricle. A small vinyl flake was passed into the ends of the
suture, which was subsequently fixed by clamping the tube with a
mosquito haemostat. Following this, the chest was closed under
negative pressure. Successful occlusion of the LAD was confirmed by
alterations in the elevation of the ST segment in Lead II and
regional cyanosis of the myocardial surface.
Assessment of infarct size
Following 4 h reperfusion, the LAD was occluded for
a second time and 2 ml Evans Blue Dye (1%; Sigma-Aldrich, St.
Louis, MO, USA) was subsequently injected via the jugular vein. The
rats were sacrificed by cardiac puncture under anesthesia with
sodium pentobarbital (45 mg/kg, i.p.). Following this, the rat
hearts were excised and frozen for 15 min prior to harvesting of
the atria and right ventricles. The left ventricles were cut into
transverse sections (thickness, 2 mm) from the apex to the base.
Subsequently, the risk areas were separated from the colored
nonischemic areas (blue), and the slices were incubated with a 1%
solution of 2,3,5-triphenyltetrazolium chloride in 0.2 M Tris
buffer (pH 7.4) stain for 15 min at 37°C. The respective infarct
sizes (white) and risk areas (red and white) of the sections were
determined using an Image-Pro Plus 3.0 image analyzer (Media
Cybernetics, Inc., Rockville, MD, USA). Infarct size was expressed
as a percentage of the risk area volume (% = infarct size/risk
area) (14).
Assessment of myocardial injury
Following 4 h reperfusion, blood samples were
collected in order to assess the levels of lactate dehydrogenase
(LDH) and creatine kinase (CK). Following a 10-min rest, blood
samples were centrifuged at 875 × g for 15 min. Prior to analyses;
the supernatants were collected and stored at −80°C. Analyses of
the LDH and CK levels were completed using commercially available
LDH and CK Assay kits and standard techniques, according to the
manufacturer's protocol (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Data are expressed in international
units (IU)/liter (l).
Assessment of myocardial inflammatory
parameters
The expression levels of TNF-α and IL-6 in the
myocardial tissue supernatants were measured using a commercial
enzyme-linked immunosorbent TNF-α assay kit (Nanjing Jiancheng
Bioengineering Institute), according to the manufacturer's
protocol. The sensitivity of the assay was 1 pg/ml for TNF-α and
IL-6.
Cardiac tissue samples were lysed in
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China) for western blotting. Protein
concentration was determined using Pierce Bicinchoninic Acid
Protein Assay kit (Pierce Biotchnology Inc., Rockville, MD, USA).
Protein extracts (50 µg) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, transferred to
polyvinylidene difluoride (PVDF) membranes (EMD Millipore,
Billerica, MA, USA) and probed with HMGB1 (1:800; cat. no. BA4277;
Wuhan Boster Biological Technology Co., Ltd., Wuhan, China), IL-17A
(1:800; cat. no. 13838; Cell Signaling Technology, Inc., Danvers,
MA, USA) and β-actin antibodies (1:800; cat. no. BM0627; Wuhan
Boster Biological Technology Co., Ltd.). Following incubation with
horseradish peroxidase-conjugated goat anti-mouse (cat. no.
BA1051), goat anti-rabbit (cat. no. BA1054) and rabbit anti-goat
(cat. no. BA1060) secondary antibodies (1:50,000; Wuhan Boster
Biological Technology Co., Ltd.) for 1 h at room temperature, the
membranes were treated with enhanced chemiluminescence reagents
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) prior to
visualization using a FluorChem E imager (ProteinSimple, San Jose,
CA, USA). The specific protein expression levels were normalized to
the levels of β-actin on the same PVDF membrane. The absorbance
values of each target protein/β-actin were used to indicate the
relative expression levels of each target protein.
Assessment of myocardial oxidative
stress
The activity levels of malondialdehyde (MDA),
superoxide dismutase (SOD), oxygen free radicals and lipid
superoxides in the myocardium were measured using commercially
available MDA and SOD Assay kits (Nanjing Jiancheng Bioengineering
Institute), according to the manufacturer's protocol.
Assessment of myocardial
apoptosis
In order to detect the apoptosis of cardiomyocytes,
terminal deoxynucleotidyl-transferase mediated dUTP nick-end
labeling (TUNEL) staining and caspase-3 activity level analyses
were performed as previously described (3). The rat hearts were fixed in 4%
paraformaldehyde, embedded in paraffin, cut into 5 µm sections and
treated as outlined in the manufacturer's protocol for the In
Situ Cell Death Detection kit (Roche Diagnostics GmbH,
Mannheim, Germany). Brown staining in the nucleus or cytoplasm
indicated the cells were TUNEL-positive, and thus apoptotic. In
order to determine the percentage of apoptotic cells, the apoptotic
cells were counted under a microscope (BX53; Olympus Corporation,
Tokyo, Japan) and the results from 10 fields of each group were
subsequently calculated.
Caspase-3 activity was measured using a caspase-3
enzyme-linked immunosorbent assay kit, according to the
manufacturer's protocol (Beyotime Institute of Biotechnology). The
absorbance of p-nitroaniline, which is cleaved by caspase-3, was
measured at 405 nm using a microplate reader (ELx800; Bio-Tek
Instruments, Winooski, VT, USA).
Statistical analysis
Unpaired Student's t-tests were used for
between-group comparisons; whereas a one-way analysis of variance
or Welch t-tests were used for comparisons among groups and a
Student-Neuman-Keuls or Dunnett T3 was used for post-hoc multiple
comparisons. Data are presented as the mean ± standard deviation.
All analyses were completed using SPSS 17.0 software (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
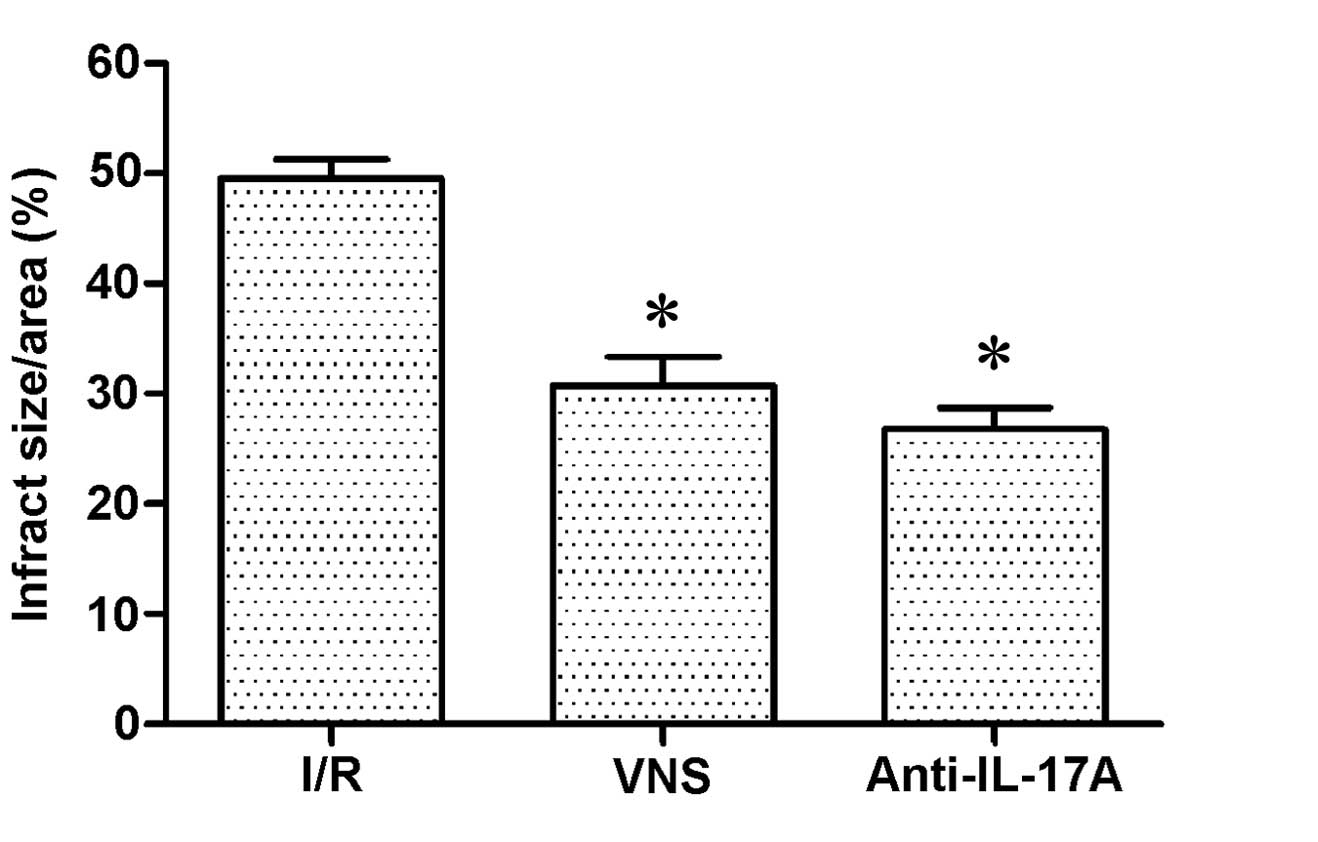
Infarct size
No evidence of myocardial infarction was detected in
the SO group, due to a lack of myocardial ischemia induction. The
infarct sizes (%) in the VS and anti-IL-17A groups were
significantly decreased, as compared with the I/R group (P<0.05;
Fig. 1)
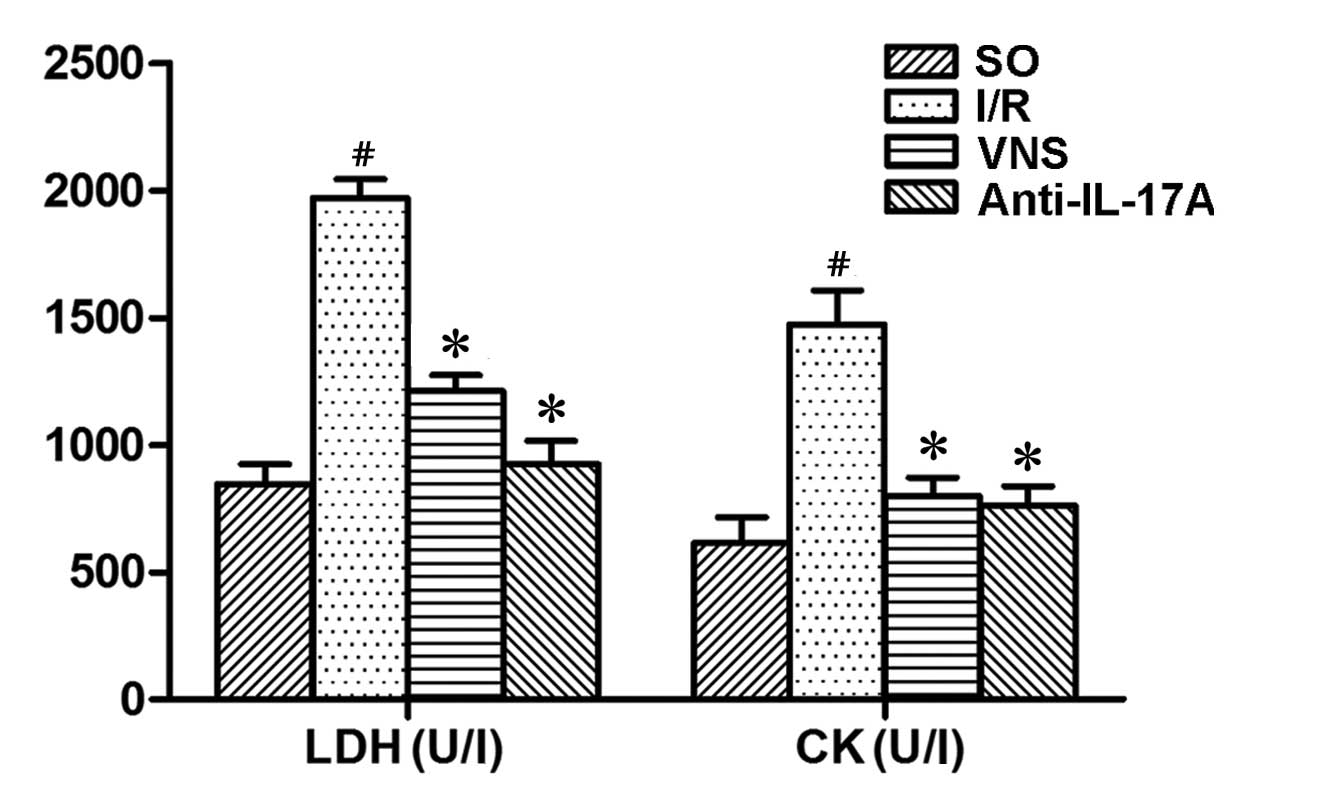
LDH and CK activity levels
Following 4 h reperfusion, the activity levels of
LDH and CK in the I/R group were significantly increased, as
compared with those in the SO group. Furthermore, VS or
administration of anti-IL-17A significantly inhibited the increase
of LDH and CK levels (P<0.05; Fig.
2)
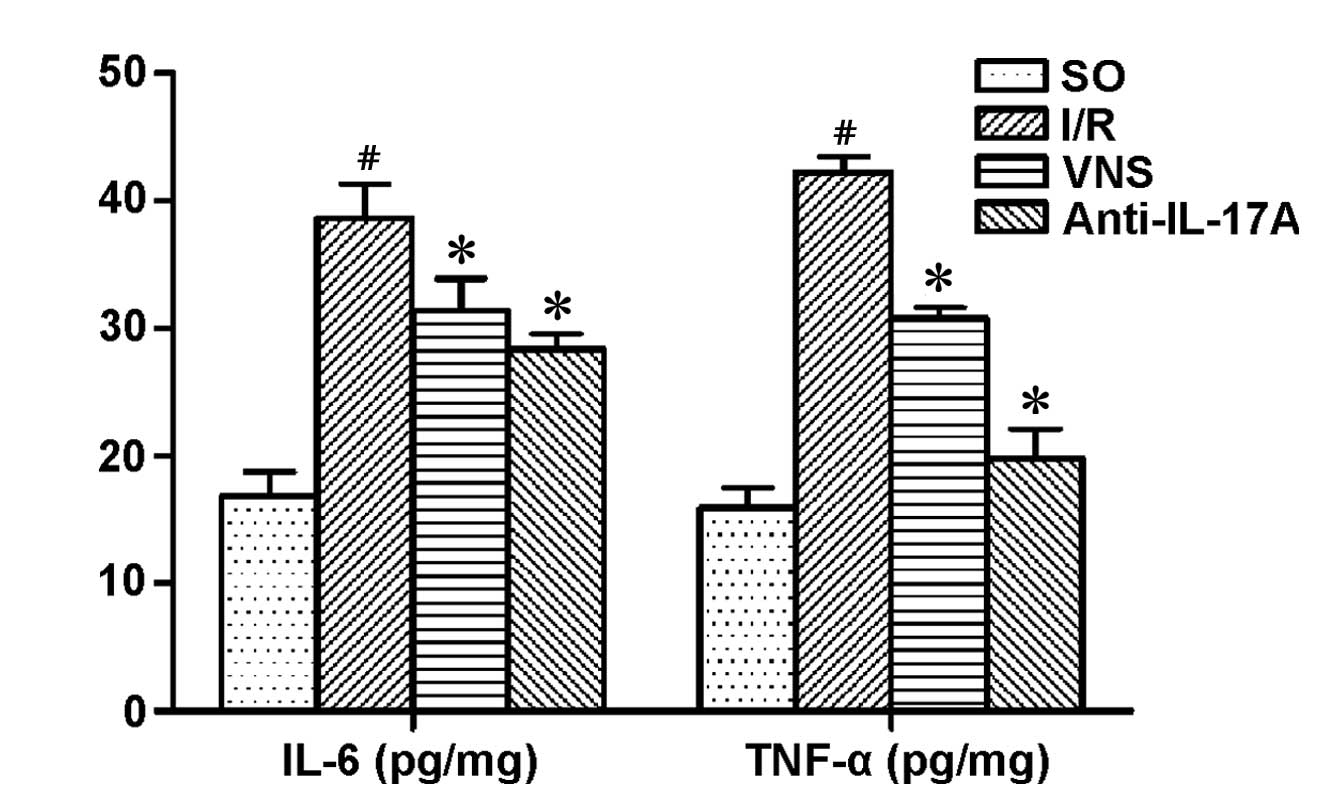
TNF-α, IL-6, IL-17A and HMGB1
expression levels
TNF-α and IL-6 levels in the I/R group were
significantly increased, as compared with the other groups
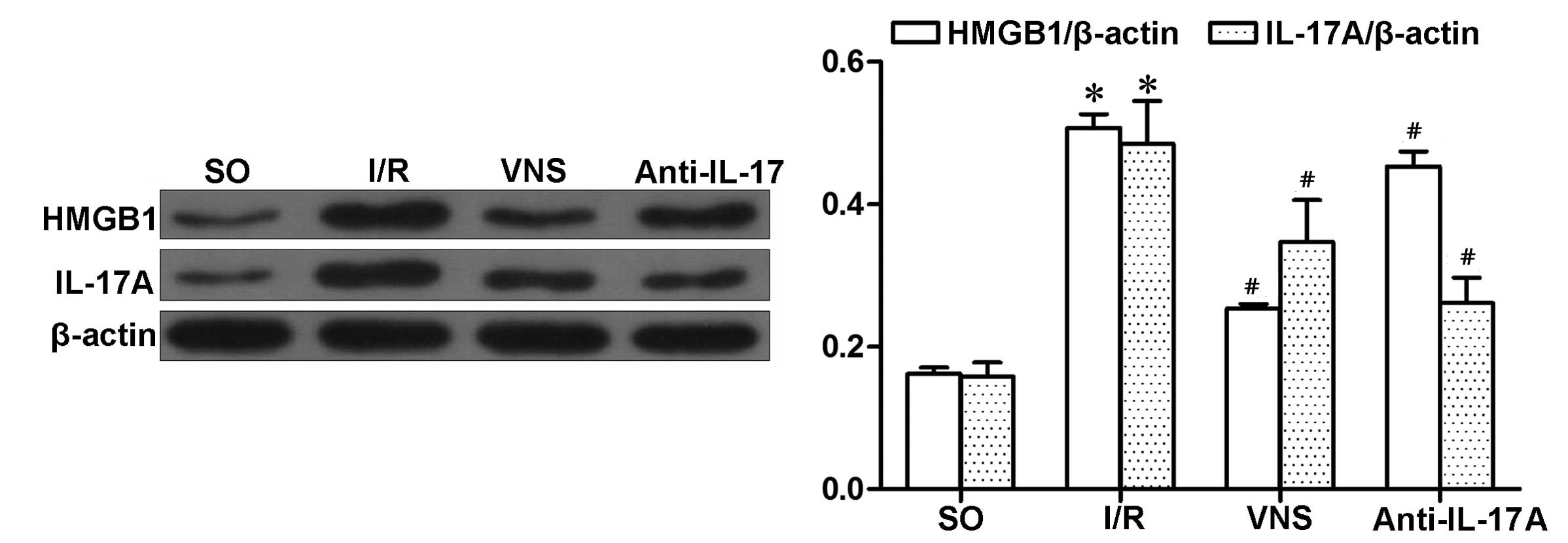
(P<0.05; Fig. 3). IL-17A and
HMGB1 expression levels in the I/R group were markedly increased,
as compared with the other groups. Similarly, VS decreased the
expression levels of IL-17A and HMGB1, as compared with the I/R
group (both P<0.05). Furthermore, administration of anti-IL-17A
mAbs inhibited the expression levels of IL-17A and HMGB1
(P<0.05; Fig. 4), which were
induced by I/R injury. However, the reductions in HMGB1 expression
levels were lower (P=0.0318; Fig.
4), as compared with in IL-17A.
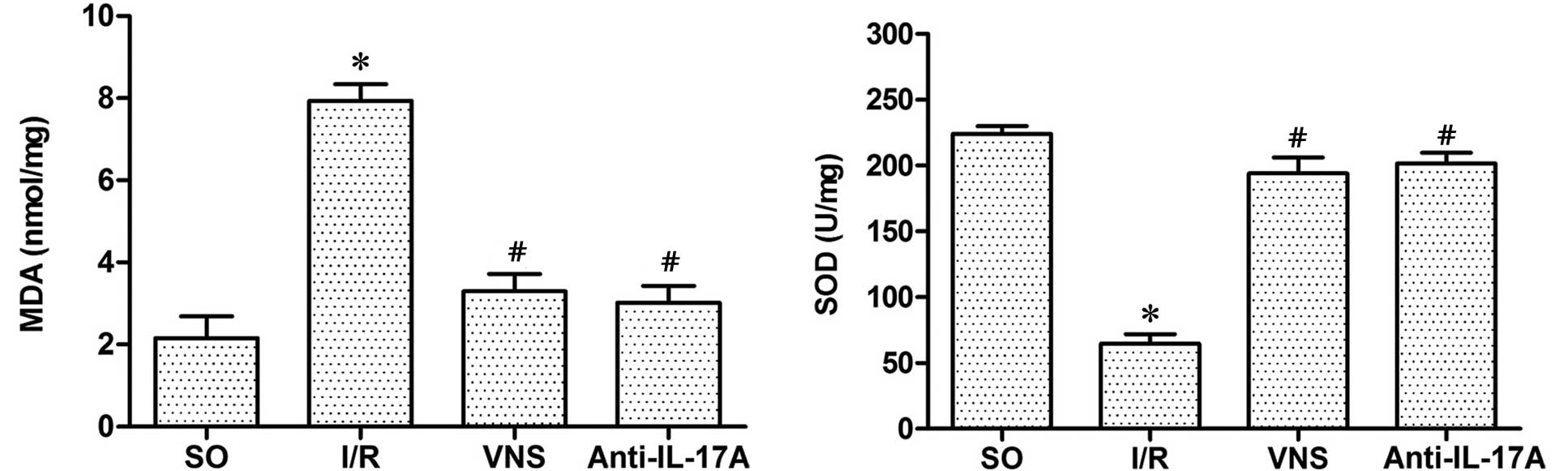
MDA and SOD activity levels
Following I/R, the levels of MDA in the I/R group
were significantly increased (P<0.05), whereas SOD levels were
significantly decreased (P<0.05), as compared with those in the
SO group. Conversely, VS or anti-IL-17A administration
significantly inhibited the oxidative stress induced by I/R
(Fig.5)
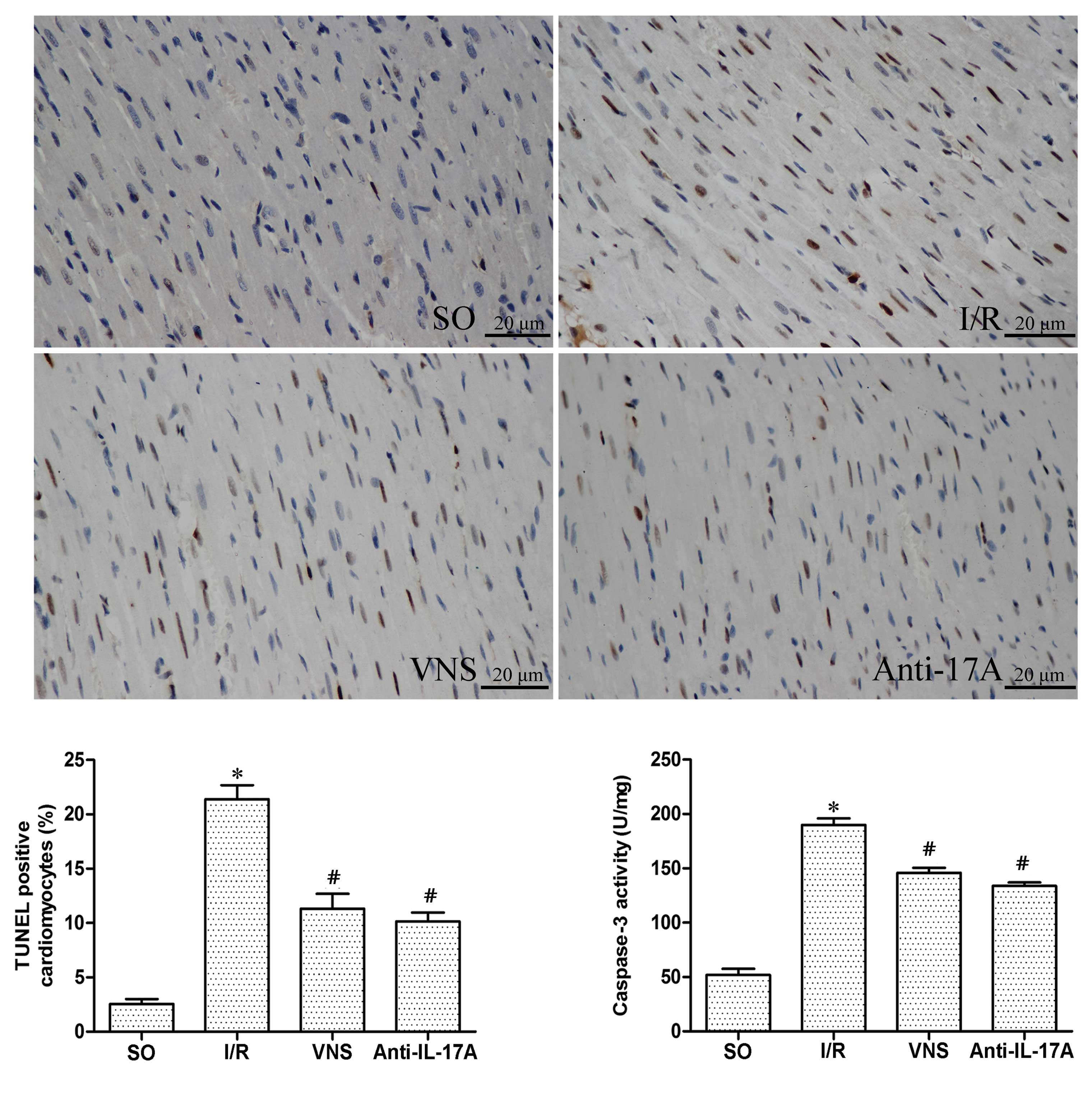
Myocardial apoptosis
The percentage of TUNEL-positive cardiomyocytes was
markedly increased in the I/R group, as compared with the other
treatment groups (Fig. 6). Similar
trends in the activity of caspase-3 were demonstrated following
I/R. These results suggest that I/R may have induced an increase in
the apoptosis of cardiomyocytes, and VS or treatment with
anti-IL-17A mAbs may attenuate I/R-induced apoptosis.
Discussion
Previous studies have demonstrated that vagal
activation is capable of exerting cardioprotective effects in
various cardiovascular diseases, including ischemic heart disease,
heart failure, arrhythmia and hypertension (7,15). In
the present study, VS was initiated at 15 min following acute
myocardial ischemia and maintained for 30 min, which is similar to
a previous study (9). Consistent
with previous studies (14,16), VS significantly reduced infarct size
following myocardial I/R injury. The results of the present study
demonstrated that VS was capable of decreasing myocardial enzyme
expression levels and cardiomyocyte apoptosis, which is an
important mechanism of cell death. Furthermore, previous studies
(16–18) have demonstrated that VS or the
neurotransmitter ACh may exert cardioprotective effects through the
regulation of mitochondrial biogenesis and function. The prolonged
opening of the mitochondrial permeability transition pore induces
cell death through the rapid depletion of ATP and activation of
death messengers, such as caspases, which have a key role in
apoptosis. The present study demonstrated that VS or anti-IL-17A
mAb treatment may regulate the apoptosis of cardiomyocytes, as
confirmed by the alterations in TUNEL-positive cardiomyocytes and
caspase-3 activity levels. These results suggested that VS may
attenuate myocardial I/R injury by restraining myocardial apoptosis
via IL-17A, which is a regulator associated with apoptosis.
Inflammation is one of the major pathophysiological
mechanisms associated with myocardial I/R injury (19). A previous study demonstrated that the
cardioprotective effects of VS in myocardial I/R injury are
associated with the activation of the α-7 nicotinic AChR-mediated
cholinergic anti-inflammatory pathway (14). HMGB1 is an integral proinflammatory
cytokine in myocardial I/R injury that communicates with other
cytokines, including TNF-α, IL-6 and C-reactive protein (20). Furthermore, α-7nAChR activation is
capable of initiating the Janus-activated kinase/signal transducers
and activators of transcription 3-nuclear factor-κB cascade, which
was associated with a decrease in the levels of proinflammatory
cytokines including TNF-α, interleukins and HMGB1 (21). Zhu et al (22) demonstrated that both HMGB1 and IL-17A
expression levels were significantly increased, and HMGB1
facilitated the release of IL-17A, which induced apoptosis and
neutrophil infiltration that further aggravated I/R injury. In the
present study, VS was able to significantly attenuate myocardial
I/R injury and decrease IL-17A, HMGB1, IL-6 and TNF-α expression
levels. Furthermore, neutralization of endogenous IL-17A
successfully protected cells against myocardial I/R injury,
inducing the same protective effects as VS; however the expression
levels of HMGB1 induced by I/R injury were only slightly reduced.
These results suggested that VS may have a cardioprotective role in
myocardial I/R injury by inhibiting HMGB1 expression levels, thus
reducing the expression levels of IL-17A.
Oxidative stress is a major detrimental process
associated with the pathogenesis of I/R injury. Ekici et al
(23) demonstrated that VS exhibits
antioxidant efficacy in the treatment of focal cerebral I/R;
therefore VS may be capable of suppressing the generation of
reactive oxygen species (ROS) in ischemic heart diseases (24,25). VS
may also exert its antioxidant effects by suppressing the prolonged
opening of the permeable transition pore which promotes the
production of ROS (16).
Furthermore, IL-17A facilitated the release of proinflammatory
cytokines and ROS, which could lead to neutrophilic inflammation
(26). Oxidative stress markers were
analyzed in the present study, the results of which demonstrated
that VS was capable of reducing the levels of MDA, a ROS, and
increasing the levels of SOD, which is a key antioxidant enzyme.
Furthermore, neutralization of endogenous IL-17A was also able to
suppress I/R-induced ROS production. These results demonstrated
that VS may alleviate myocardial I/R injury by suppressing the
I/R-induced production of ROS, which may be associated with the
suppression of IL-17A expression levels.
In the present study, VS intervention was performed
as previously described (9). The
time schedule of treatment in the present study may differ from the
clinical condition. Furthermore, VS and anti-IL-17A mAb treatment
demonstrated antioxidant effects and the ability to inhibit
apoptosis in cardiomyocytes; thus suggesting that IL-17A may be a
downstream regulator of the cholinergic anti-inflammatory pathway
activated by VS. Investigation of the VS-HMGB1-IL-17A axis remains
necessary, in order to directly understand the relationship between
VS and IL-17A.
In conclusion, the present study demonstrated that,
in a rat model of myocardial I/R, VS is capable of modulating
inflammatory responses and inhibiting oxidative stress and
apoptosis-induced injury. Furthermore, VS may attenuate myocardial
I/R injury, which may be associated with the inhibition of IL-17A
expression.
Acknowledgements
The present study was partially supported by a grant
from the National Natural Science foundation of China (grant no.
81370308).
References
|
1
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iwakura Y, Nakae S, Saijo S and Ishigame
H: The roles of IL-17A in inflammatory immune responses and host
defense against pathogens. Immunol Rev. 226:57–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ni J, Hu G, Xiong J, Shen J, Shen J, Yang
L, Tang M, Zhao Y, Ying G, Yu G, et al: Involvement of
interleukin-17A in pancreatic damage in rat experimental acute
necrotizing pancreatitis. Inflammation. 36:53–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Sun R, Wei H and Tian Z:
High-mobility group box 1 (HMGB1)-Toll-like receptor
(TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced
damage-associated lethal hepatitis: Interaction of γδ T cells with
macrophages. Hepatology. 57:373–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu X, Xu W and Jiang H: HMGB1/IL-17A axis:
An important mechanism for myocardial ischemia-reperfusion injury.
Int J Cardiol. 174:447–448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX,
Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, et al: Interleukin-17A
contributes to myocardial ischemia/reperfusion injury by regulating
cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll
Cardiol. 59:420–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao M, Sun L, Liu JJ, Wang H, Miao Y and
Zang WJ: Vagal nerve modulation: A promising new therapeutic
approach for cardiovascular diseases. Clin Exp Pharmacol Physiol.
39:701–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Ferrari GM and Schwartz PJ: Vagus nerve
stimulation: from pre-clinical to clinical application: challenges
and future directions. Heart Fail Rev. 16:195–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Q, Cheng Y, Xue FS, Yuan YJ, Xiong J,
Li RP, Liao X and Liu JH: Postconditioning with vagal stimulation
attenuates local and systemic inflammatory responses to myocardial
ischemia reperfusion injury in rats. Inflamm Res. 61:1273–1282.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park J, Kang JW and Lee SM: Activation of
the cholinergic anti-inflammatory pathway by nicotine attenuates
hepatic ischemia/reperfusion injury via heme oxygenase-1 induction.
Eur J Pharmacol. 707:61–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang Q, Li J, Zhu H, Li P, Zou Z and Xiao
Y: Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in
murine models of melanoma. Mediators Inflamm. 2013:7138592013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Calvillo L, Vanoli E, Andreoli E, Besana
A, Omodeo E, Gnecchi M, Zerbi P, Vago G, Busca G and Schwartz PJ:
Vagal stimulation, through its nicotinic action, limits infarct
size and the inflammatory response to myocardial ischemia and
reperfusion. J Cardiovasc Pharmacol. 58:500–507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
National Research Council; Guide for the
Care and Use of Laboratory Animals. Washington (DC): National
Academies Press (US). 1996.
|
|
14
|
Hu X, Zhou X, He B, Xu C, Wu L, Cui B, Wen
H, Lu Z and Jiang H: Minocycline protects against myocardial
ischemia and reperfusion injury by inhibiting high mobility group
box 1 protein in rats. Eur J Pharmacol. 638:84–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thayer JF and Lane RD: The role of vagal
function in the risk for cardiovascular disease and mortality. Biol
Psychol. 74:224–242. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shinlapawittayatorn K, Chinda K, Palee S,
Surinkaew S, Thunsiri K, Weerateerangkul P, Chattipakorn S,
KenKnight BH and Chattipakorn N: Low-amplitude, left vagus nerve
stimulation significantly attenuates ventricular dysfunction and
infarct size through prevention of mitochondrial dysfunction during
acute ischemia-reperfusion injury. Heart Rhythm. 10:1700–1707.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katare RG, Ando M, Kakinuma Y, Arikawa M,
Handa T, Yamasaki F and Sato T: Vagal nerve stimulation prevents
reperfusion injury through inhibition of opening of mitochondrial
permeability transition pore independent of the bradycardiac
effect. J Thorac Cardiovasc Surg. 137:223–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK
and Zang WJ: Cardioprotection by acetylcholine: A novel mechanism
via mitochondrial biogenesis and function involving the PGC-1α
pathway. J Cell Physiol. 228:1238–1248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion - from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu X, Fu W and Jiang H: HMGB1: A potential
therapeutic target for myocardial ischemia and reperfusion injury.
Int J Cardiol. 155:4892012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marrero MB and Bencherif M: Convergence of
alpha 7 nicotinic acetylcholine receptor-activated pathways for
anti-apoptosis and anti-inflammation: Central role for JAK2
activation of STAT3 and NF-kappaB. Brain Res. 1256:1–7. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu H, Li J, Wang S, Liu K, Wang L and
Huang L: Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion
injury in a cardiac transplantation model. Transplantation.
95:1448–1454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ekici F, Karson A, Dillioglugil MO, Gurol
G, Kir HM and Ates N: The effects of vagal nerve stimulation in
focal cerebral ischemia and reperfusion model. Turk Neurosurg.
23:451–457. 2013.PubMed/NCBI
|
|
24
|
Tsutsumi T, Ide T, Yamato M, Kudou W,
Andou M, Hirooka Y, Utsumi H, Tsutsui H and Sunagawa K: Modulation
of the myocardial redox state by vagal nerve stimulation after
experimental myocardial infarction. Cardiovasc Res. 77:713–721.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kong SS, Liu JJ, Yu XJ, Lu Y and Zang WJ:
Protection against ischemia-induced oxidative stress conferred by
vagal stimulation in the rat heart: Involvement of the AMPK-PKC
pathway. Int J Mol Sci. 13:14311–14325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pinart M, Zhang M, Li F, Hussain F, Zhu J,
Wiegman C, Ryffel B and Chung KF: IL-17A modulates oxidant
stress-induced airway hyperresponsiveness but not emphysema. PLoS
One. 8:e584522013. View Article : Google Scholar : PubMed/NCBI
|