Introduction
Amyloidosis was first described in 1854 by the
pathologist Rudolph Virchow, and is a systemic disease that results
from the tissue deposition of misfolded proteins, causing the
progressive failure of organs (1). A
total of 6 types of amyloidosis exist; these are primary,
secondary, hemodialysis-associated, hereditary, senile and
localized (2). The most common of
these 6 types is primary amyloidosis, the prevalence of which is
~4.5 in 10,000 cases (3). Primary
amyloidosis, in which fibrils are made up of a monoclonal
immunoglobulin light chain, is the most common and the most severe
form of amyloidosis. The present study presents a case of primary
amyloidosis with digestive system involvement in the form of
abdominal distension and diarrhea.
Case report
A 51-year-old male patient was referred to the
Department of Hepatopancreatobiliary Surgery, The Fifth Affiliated
Hospital of Wenzhou Medical University (Lishui, Zhejiang, China) on
March 5, 2014 with a 5-month history of abdominal distension and
diarrhea, excreting yellow, loose stool 4–5 times a day that
contained no pus, blood or mucus. The patient presented with no
abdominal pain, acid reflux, chest tightness, difficulty breathing,
chills or fever, and was otherwise healthy, indicating no signs of
osteomyelitis, myeloma, rheumatoid arthritis or an associated
family history of these conditions.
A physical examination revealed no significant
abnormalities; the patient was negative for macroglossia (Fig. 1), which is indicative of
gastrointestinal tract involvement, and results for the complete
blood count, echocardiogram, erythrocyte sedimentation rate, and
basic metabolic and liver function tests were all normal. However,
serum and urine protein electrophoresis tests indicated an abnormal
κ/λ ratio of 0.7 (normal range, 1.5–3.0). B-mode ultrasonography
demonstrated changes to the kidneys (enhanced enchogenicity and
renal parenchyma) and ascites; gastroscopy also demonstrated an
ulcerous duodenal bulb (S1 gastric ulcer stage) and superficial
gastritis. A needle biopsy and subsequent microscopic evaluation of
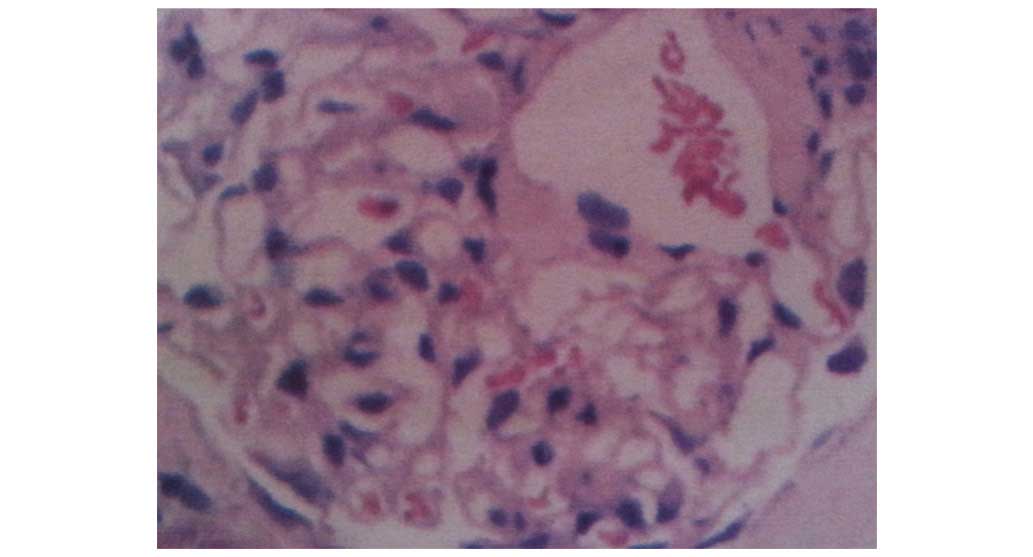
the kidney revealed Congo red-positive material deposited within
the mesangium and renal interstitium (Fig. 2), enabling a histopathological
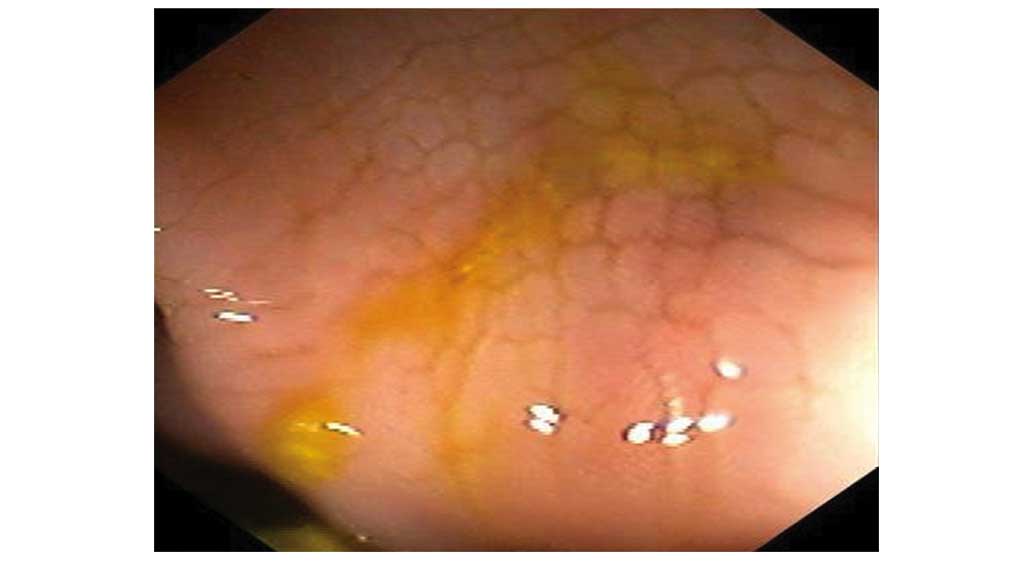
diagnosis of renal amyloidosis. A colonoscopy revealed
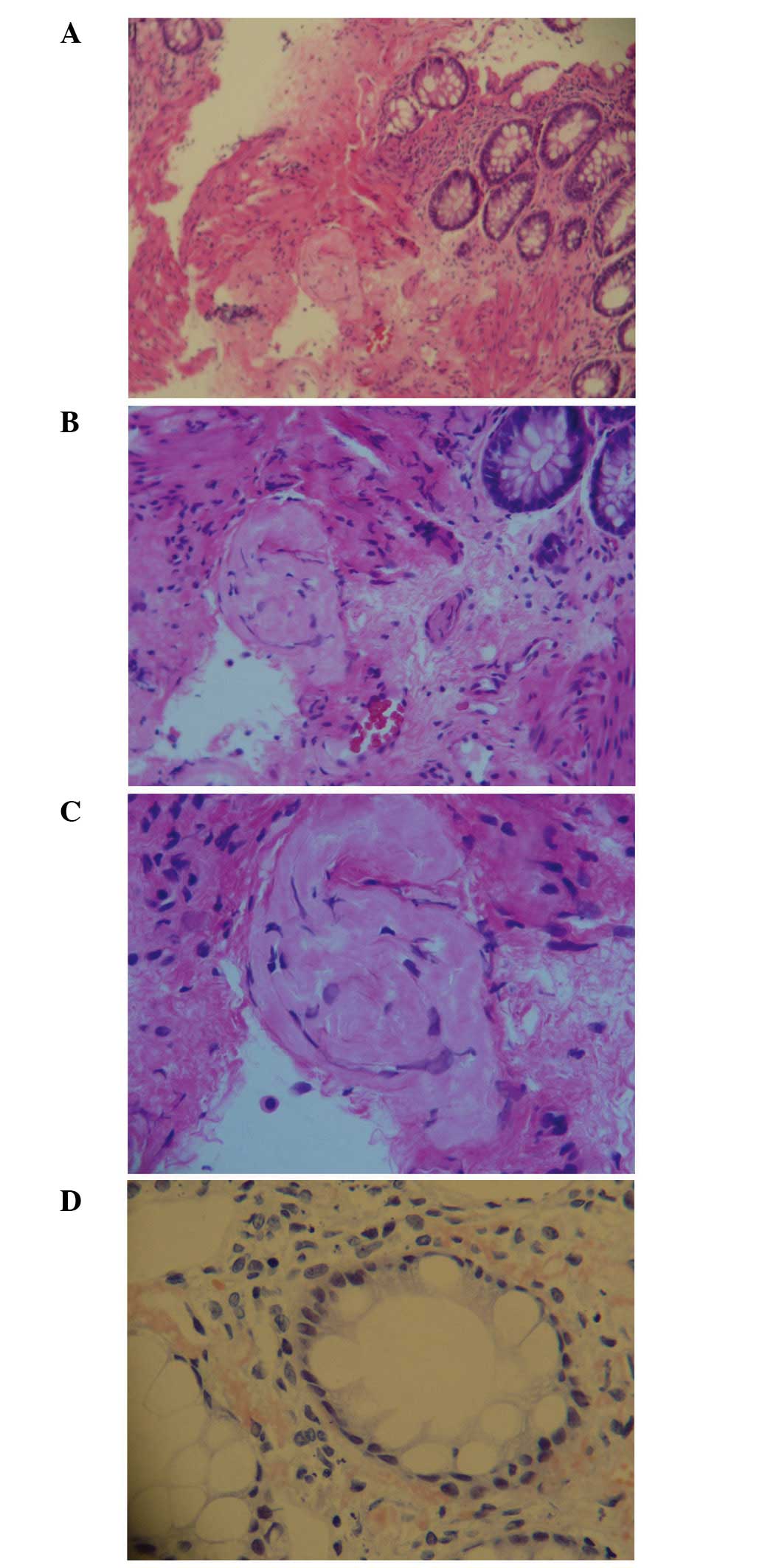
‘cobblestone’ patterns of ulcerations in the colon (Fig. 3), and the interstitium of the
mucoderm and submucosa surrounding the blood vessels of the rectal
tissues demonstrated positive staining for Congo red (Fig. 4). From the integration of these
physical, laboratory and histological examinations, a diagnosis of
localized primary amyloidosis was formed.
The patient was initially diagnosed with nephrosis,
duodenal bulb ulcers and functional enteritis prior to amyloidosis.
Following a diagnosis, the patient was treated with bifidobacteria
(1 g tid PO) to modulate the intestinal microflora, α-keto acid
tablets (2.52 g tid PO) to protect the kidneys by reducing the
synthesis of urea and the accumulation of toxic uremic products,
omeprazole enteric-coated capsules (20 mg qd PO) to protect the
gastrointestinal tract from acidic damage, and enteritis syrup (10
ml tid PO) and composite Lactobacillus capsules (0.33 g tid
PO) to treat the diarrhea. Following treatment with these drugs,
there was no marked improvement of symptoms, and the patient
declined the continuation of treatment.
Discussion
Amyloidosis is a rare disease, and its prognoses and
clinical manifestations are broad. Common symptoms of primary
amyloidosis include fatigue, dyspnoea and frequently, kidney
disease; patients often develop proteinuria, which progresses into
renal failure (4). Cardiac
involvement is also prevalent; heart failure and arrhythmia are the
predominant cause of mortality from amyloidosis (5). Involvement of other organs occurs as
follows: Hepatic deposition causes liver enlargement and increased
alkaline phosphatase levels; depositions on the spleen or lungs
leads to impairment of spleen function or respiratory failure,
respectively; and cutaneous depositions trigger the formation of
bilateral periorbital purpura, papules and plaques (6). Macroglossia typically occurs as a
result of oral lesions, and leads to sleep apnea and dysphasia. The
clinical manifestations of gastrointestinal involvement are
determined by the location and quantity of protein deposition.
Gastric and duodenal involvement can cause symptoms including
nausea, abdominal pain and hematemesis, and small intestinal
involvement can result in symptoms such as diarrhea, steatorrhea,
gastrointestinal bleeding and obstruction.
The diagnosis of amyloidosis requires a
combinatorial approach of clinical and imaging examinations and
pathological diagnosis. Previous studies have revealed that
positron emission tomography/computed tomography (CT) using
18F-fluorodeoxyglucose (18F-FDG) as an
imaging agent can be used to detect the lung nodules of amyloid
deposition, as these nodules tend to absorb FDG (7), and increased 18F-FDG uptake
is also reported in the livers of patients with primary amyloidosis
(8). Hypoperfusion of the spleen,
demonstrated using an enhanced CT scan, may also enable the
diagnosis of systemic amyloidosis (9). All the aforementioned non-invasive
techniques aid the diagnosis of amyloidosis, but the pathological
examination of organs, such as through use of Congo red staining of
the biopsy, remains the most effective way to confirm this
diagnosis (10).
There are no specific treatments for amyloidosis at
present, the aims of which would be to eliminate the light chain of
the misfolding amyloid protein, and to support the function of
affected organs. Dexamethasone has replaced prednisone in
combination treatments with melphalan due to the greater
hematological response to this regimen (11). Intravenous administration of
high-dose melphalan combined with autologous hematopoietic stem
cell transplantation can also improve the patient survival rate
(11). Due to the broad nature of
symptoms, individualized treatments of the target organs may also
be used; continuous peritoneal dialysis and kidney transplants are
useful in renal lesions, for instance (12), and diuretics can be used to treat
patients with congestive heart failure (13).
The misdiagnosis rate of amyloidosis is high, due to
the fact that the clinical manifestations of primary amyloidosis
are complex, varied, and lack specificity, which often delays
diagnosis. In this present case, abdominal distension and diarrhea
as the primary symptoms, although these are not common symptoms. In
clinical settings disease symptoms should be analyzed
systematically, and rare diseases should be considered following
the exclusion of common diseases to avoid delays in diagnosis.
References
|
1
|
Sipe JD and Cohen AS: Review: History of
the amyloid fibril. J Struct Biol. 130:88–98. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bhat A, Selmi C, Naguwa SM, Cheema GS and
Gershwin ME: Currents concepts on the immunopathology of
amyloidosis. Clin Rev Allergy Immunol. 38:97–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gertz MA, Lacy MQ and Dispenzieri A:
Amyloidosis: Recognition, confirmation, prognosis, and therapy.
Mayo Clin Proc. 74:490–494. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anupama YJ and Vankalakunti M: Rapidly
progressive glomerulonephritis in a patient with renal amyloidosis:
Case report and review of the literature. Indian J Nephrol.
22:377–380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gertz MA, Comenzo R, Falk RH, Fermand JP,
Hazenberg BP, Hawkins PN, Merlini G, Moreau P, Ronco P,
Sanchorawala V, et al: Definition of organ involvement and
treatment response in immunoglobulin light chain amyloidosis (AL):
A consensus opinion from the 10th International Symposium on
Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J
Hematol. 79:319–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Desport E, Bridoux F, Sirac C, Delbes S,
Bender S, Fernandez B, Quellard N, Lacombe C, Goujon JM, Lavergne
D, et al: Centre national de référence pour l'amylose AL et les
autres maladies par dépôts d'immunoglobulines monoclonales: Al
amyloidosis. Orphanet J Rare Dis. 7:542012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seo JH, Lee SW, Ahn BC and Lee J:
Pulmonary amyloidosis mimicking multiple metastatic lesions on F-18
FDG PET/CT. Lung Cancer. 67:376–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Son YM, Choi JY, Bak CH, Cheon M, Kim YE,
Lee KH and Kim BT: 18F-FDG PET/CT in primary AL hepatic amyloidosis
associated with multiple myeloma. Korean J Radiol. 12:634–637.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mainenti PP: RE: Imaging features of
hepato-splenic amyloidosis at PET/CT. Korean J Radiol. 13:368–369.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki K: Diagnosis and treatment of
multiple myeloma and AL amyloidosis with focus on improvement of
renal lesion. Clin Exp Nephrol. 16:659–671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Merlini G, Seldin DC and Gertz MA:
Amyloidosis: Pathogenesis and new therapeutic options. J Clin
Oncol. 29:1924–1933. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gude D, Chennemsetty S, Jha R and Narayan
G: Primary amyloidosis treated with continuous ambulatory
peritoneal dialysis. Saudi J Kidney Dis Transpl. 23:1285–1287.
2012.PubMed/NCBI
|
|
13
|
Falk RH: Cardiac amyloidosis: A treatable
disease, often overlooked. Circulation. 124:1079–1085. 2011.
View Article : Google Scholar : PubMed/NCBI
|