Introduction
Cell-penetrating peptides (CPPs), also known as
protein transduction domains (1) or
Trojan peptides, are able to overcome limitations of large
molecular (DNA and proteins) drug delivery systems (viral and
non-viral) commonly used currently, including membrane penetration
and toxicity (2). CPPs
non-invasively translocate through the cell membrane to
ubiquitously deliver a wide-range of hydrophilic and hydrophobic
bioactive molecules, including proteins, DNA and liposomes; thus,
CPPs can be used as a versatile tool for intracellular delivery
(3). Although the exact mechanisms
underlying CPP internalization are unclear, it is speculated that
various endocytic pathways and direct membrane translocation are
involved (4).
The tegument protein VP22 of the Herpes simplex
virus type 1 (HSV-1) is a naturally occurring CPP with an effective
intracellular spread (5,6). The recombinant VP22 protein augments
the therapeutic effects of protein drugs (7), as well as of gene and immune therapies
(8,9), by increasing the intracellular
distribution of therapeutic molecules. VP22 fusion-proteins are
currently in use for the treatment of diseases such as congestive
heart failure and C6 glioma (8,10). The
aim of the present study was to provide novel molecular tools to
assist advancement in the development of VP22-fusion therapeutics.
Thus, the expression of peptides corresponding to the N-terminus
and C-terminus of VP22 were detected, and subsequently antisera
against these peptides were produced, which detected both the
truncated and the full-length VP22 protein.
Materials and methods
Plasmids and strains
pcDNA3-VP22 expressing HSV 1 VP22 was constructed as
described previously (11).
Glutathione S-transferase (GST)-tagged pGEX-5X-1 (GE Healthcare
Life Sciences, Little Chalfont, UK) vector acted as prokaryotic
expression vectors. E. coli BL21 (DE3) cells (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) were used for
inducible protein expression.
Cells
Vero cells (Laboratory of Biochemistry and Molecular
Pharmacology, Chongqing Medical University, Chongqing, China) were
used for verification of polyclonal antibody binding.
Experimental animals
A total of 20 female BALB/c mice (age, 4–6 weeks;
weight, 15 g; Animal Laboratory Center of Chongqing Medical
University) were maintained in specific pathogen-free,
environmentally controlled conditions at 22±2°C with 50–70%
humidity. Animals had ad libitum access to food and water.
The use of animals and the experimental protocols were approved by
the Ethics Committee of Chongqing Medical University.
Cloning of VP22 peptides into the
pGEX-5X-1 vector
A pcDNA3-VP22 containing the HSV-1 VP22 gene was
constructed as described previously (11). Briefly, the HSV-1 VP22 gene was
amplified using polymerase chain reaction (PCR) from a HSV-1 virus
harvest. The VP22 amplicon was digested with EcoRI (Takara
Biotechnology Co., Ltd., Dalian, China) and subcloned into a
eukaryotic expression vector pcDNA3 (Invitrogen; Thermo Fisher
Scientific, Inc.), resulting in pcDNA3-VP22. Using pcDNA3-VP22 as a
template, the DNA sequence expressing the first 60 amino acids (aa)
of the N-terminus and the last 45 aa of the C-terminus of VP22 were
amplified by PCR. The primers and restriction enzymes used for
recombination are listed in Table I.
PCR was performed on GeneAmp 2400 PCR system (PerkinElmer, Boston,
MA, USA), with an initial denaturation at 94°C for 3 min, followed
by 30 cycles of denaturation at 94°C for 30 sec, annealing at 55°C
for 30 sec, extension at 72°C for 30 sec, and a final extension at
72°C for 10 min. The amplified genes were gel-purified using an
agarose gel extraction kit (Tiangen Biotech Co., Ltd., Beijing,
China), according to the manufacturer's instructions. Double
restriction endonuclease digestion using EcoRI/XhoI
(Takara Biotechnology Co., Ltd.) was performed in pGEX-5X-1 and the
PCR fragment expressing N-terminal 60 aa of VP22. Double
restriction endonuclease digestion using BamHI/EcoRI
(Takara Biotechnology Co., Ltd.) was performed in pGEX-5X-1 and the
PCR fragment expressing the C-terminal 45 aa of VP22. The digested
target fragments and vectors were ligated at 16°C for 1 h using a
T4 DNA ligase kit (Takara Biotechnology Co., Ltd.) resulting in
pGEX-N60 (expressing GST-tagged N-terminal 60 aa of VP22) or
pGEX-C45 (expressing GST-tagged C-terminal 45 aa of VP22). The
recombinant vectors were identified by restriction analysis with
EcoRI/XhoI or BamHI/EcoRI at 37°C for 2 h and DNA sequencing by
Sangon Biotech Co., Ltd. (Shanghai, China), after which they were
transformed into competent E. coli BL21 (DE3) cells.
 | Table I.Primers for the construction of
recombinant plasmids. |
Table I.
Primers for the construction of
recombinant plasmids.
| Primer | Restriction site | Sequence (5′ to
3′) |
|---|
| N-terminal 60 aa
forward | EcoRI |
ATAGAATTCATGACCTCTCGCCGC |
| N-terminal 60 aa
reverse | XhoI |
ATTCTCGAGGTACTGGACGAAACG |
| C-terminal 45 aa
forward | BamHI |
ATCGGATCCAAGAGTTGGTGAATCCA |
| C-terminal 45 aa
reverse | EcoRI |
ATTGAATTCTCACTCGACGGGCCG |
Expression of recombinant VP22
proteins
E. coli BL21 (DE3) cells were chemically
transformed with pGEX-N60 or pGEX-C45, and grown overnight in
Luria-Bertani medium (Sigma-Aldrich, St. Louis, MO, USA) containing
100 µg/ml ampicillin (Tiangen Biotech Co., Ltd.) at 37°C. Next, 0.5
ml of the overnight E. coli cell culture was transferred into fresh
medium in a culture flask and grown until an optical density at 600
nm of 0.5 was reached (SP-756 UV-Vis Spectrophotometer; Shanghai
Spectrum Instrument Co., Ltd., Shanghai, China). Subsequently,
expression of the recombinant protein was induced by addition of 1
mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 4 h at
25°C.
Extraction of recombinant VP22
protein
E. coli BL21 (DE3) expressing cells were
collected by centrifugation at 4,000 × g for 15 min at room
temperature. The cell pellet was washed three times with
double-distilled (dd)H2O and incubated in 5 ml lysis
buffer [50 mM phosphate-buffered saline (PBS), pH 7.4; 0.5 M NaCl;
1 mM MgCl2; 0.5 mg/ml lysozyme; and 1 mM
phenylmethylsulfonyl fluoride] on ice for 45 min. Next, the cells
were sonicated at 50% duty cycle and 300 W for 8 min using an
ultrasonic disintegrator (scientz-IID; Ningbo Xinzhi Instruments
Inc., Ningbo, China). The soluble fraction was then collected
following centrifugation at 15,000 × g for 10 min at 4°C. The
inclusion bodies (insoluble fraction) were dissolved in 2 ml urea
(6 M), with incubation at 42°C for 30 min, and then recovered by
centrifugation at 8,000 × g for 10 min at room temperature.
Subsequently, the soluble fraction and the dissolved inclusion
bodies were subjected to 12% SDS-PAGE and Coomassie Brilliant Blue
R250 (Beyotime Institute of Biotechnology, Haimen, China) staining
to determine the protein expression.
Purification of recombinant VP22
proteins by electroelution
The excised recombinant protein bands were subjected
to electroelution at 4°C for 3 h at 100 mA, using an electroelution
buffer (25 mM Tris-HCl, 250 mM glycine and 0.1% SDS; pH 8.3) and a
dialysis bag (Sangon Biotech Co., Ltd.) with a 6-kDa molecular
weight cut-off. Electroelution was terminated when the Coomassie
Brilliant Blue R250 dye completely ran from the SDS-PAGE gels into
electroelution completely, and the electroelution was incubated in
five volumes of acetone at −20°C for 1 h. The recombinant protein
pellet was harvested by centrifugation at 12,000 × g for 20 min at
4°C, dissolved in ddH2O and desalted by running it
through a Sephadex G-25 column (GE Healthcare Life Sciences) to
remove excess small molecules (12).
Production and purification of
polyclonal antisera against the recombinant proteins
Antibodies against each recombinant VP22 protein
were obtained by immunizing 4–6-week-old female BALB/c mice. Each
animal was initially injected subcutaneously with 50 µg purified
recombinant protein emulsified in Freund's complete adjuvant (1:1;
Sigma-Aldrich), subsequent to collecting pre-immune sera. Two
booster injections of 0.5 mg/ml recombinant protein in equal volume
of incomplete Freund's adjuvant (Sigma-Aldrich) were given at
2-week intervals in order to obtain a prolonged persistence of the
immunogen in the tissues and a continuous stimulation of the immune
system. At 10 days after the final injection, blood was collected
from the tail vein of the immunized mice, and the crude antisera
were collected by centrifugation at 4,200 × g for 5 min at room
temperature. The immunoglobulin (Ig) antibodies were precipitated
with ammonium sulfate (40% saturation) and dissolved in 2 ml of 0.2
M NaCl.
ELISA validation of the polyclonal
antisera
ELISA plates (Corning, Inc., Corning, NY, USA) were
coated with 100 µl purified recombinant VP22 proteins (5 µg/ml) in
50 mM carbonate-bicarbonate buffer (pH 9.6) and incubated at 4°C
overnight. Subsequent to washing with 0.5% PBS-Tween-20 (pH 7.6),
non-specific proteins were blocked with 150 µl 5% bovine serum
albumin for 1 h at room temperature and then incubated with 100 µl
polyclonal antiserum of different dilutions (between 1:200 and
1:64,000) for 2 h at 37°C. Following incubation with 100 µl of
horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1:2,000; sc-2005)
for 1 h at 37°C, the proteins were detected using a TMB Detection
system (Wuhan Boster Biological Technology, Ltd., Wuhan, China),
and binding was quantified by measuring the absorbance at 450 nm
with SpectraMax 190 microplate reader (Molecular Devices LLC,
Sunnyvale, CA, USA). The pre-immunized mouse serum served as a
negative control.
Western blot analysis
Vero cells grown to 70% confluence in 6-well plates
and were transfected with pcDNA3-VP22 (the full length VP22),
pcDNA3-VP22-N (the first 150 aa of the VP22 N-terminus), or
pcDNA3-VP22-C (the last 151 aa of the VP22 C-terminus). After 48 h,
the cells were lysed in radioimmunoprecipitation assay buffer
(Bryotime Institute of Biotechnology), and the proteins were
subjected to 12% SDS-PAGE and then electrotransferred to a
polyvinylidene difluoride membrane. Incubation with the purified
anti-recombinant VP22 (1:500) was followed by incubation with
secondary HRP-conjugated goat anti-mouse IgG (Santa Cruz
Biotechnology, Inc.; 1:2,000; sc-2005), and the proteins were
detected using the DAB Detection Kit (Wuhan Boster Biological
Technology, Ltd.). The empty pcDNA3-transfected cells served as a
negative control.
Immunofluorescence assay
Vero cells were grown to 70% confluence on glass
coverslips and transfected with the pcDNA3-VP22 plasmid. At 48 h
post-transfection, the cells were washed with PBS, fixed in cold
methanol for 10 min at room temperature, and then permeabilized
with 0.2% Triton X-100-PBS (Sigma-Aldrich) for 90 min at room
temperature. Following a wash with PBS, the cells were blocked for
30 min in 5% non-fat milk at room temperature and incubated with
anti-recombinant VP22 sera (1:100) at 4°C overnight. Subsequent to
three further washes with PBS, the cells were incubated with
fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Santa
Cruz Biotechnology, Inc.; 1:1,000; sc-2010) for 1 h at room
temperature and observed by inverted fluorescence microscopy (TCS
SP2; Leica Microsystems, Wetzlar, Germany). The pre-immunized mouse
serum served as negative control.
Results
Expression vector construction
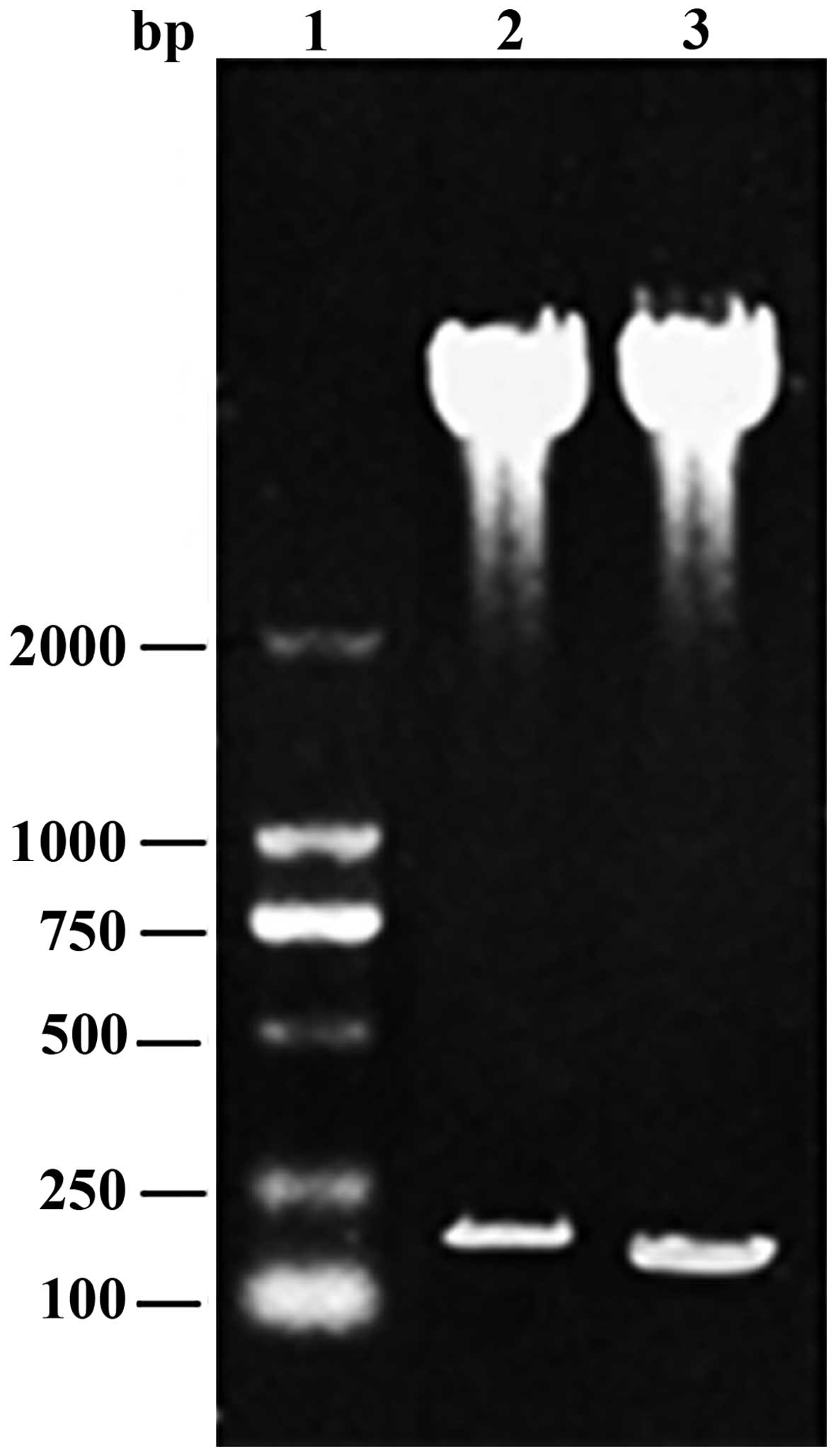
The PCR-amplified N-terminal (60 aa) and C-terminal
(45 aa) DNA sequences corresponded with the expected sizes of 180
bp and 135 bp, respectively. The PCR products were ligated into the
pGEX-5X-1 vector and transformed into competent E. coli BL21 (DE3)
cells, and the positive clones were identified by restriction
analysis (Fig. 1). DNA sequencing
confirmed that pGEX-N60 and pGEX-C45 were constructed.
Expression and purification of the
recombinant proteins
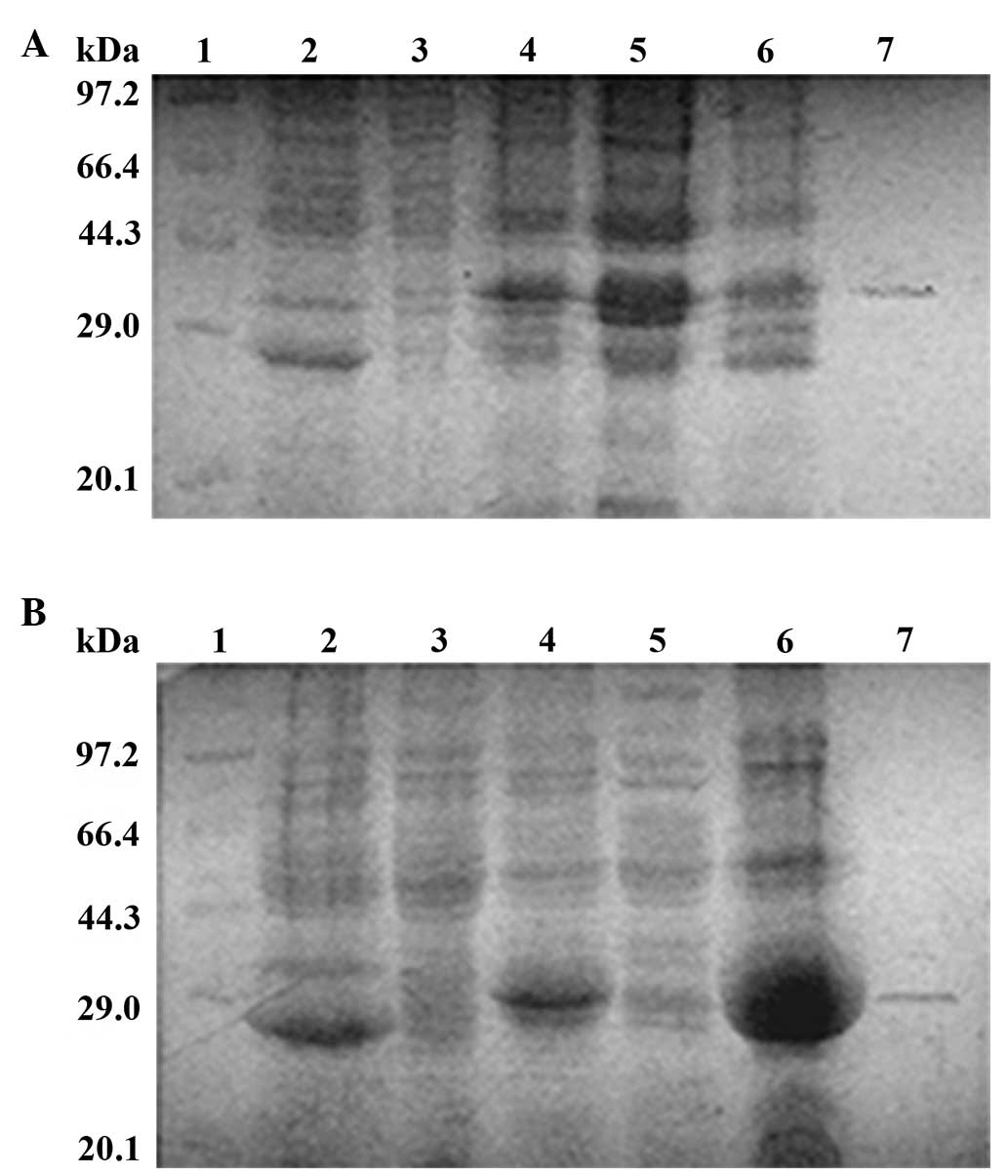
IPTG addition induced the expression of recombinant
proteins in the E. coli BL21 (DE3) transformants. Since the
molecular weight of GST is 26 kDa, the expected molecular weights
for GST-tagged N-terminal 60 aa of VP22 (N60-GST) and GST-tagged
C-terminal 45 aa of VP22 (C45-GST) were 32 kDa and 30 kDa,
respectively. N60-GST and C45-GST migrated to a position in the gel
close to the 29 kDa band in the molecular weight markers and were
close to the expected molecular weights (Fig. 2A and B, lanes 4–6). In addition, the
target protein was almost absent in the non-induced transformants
(Fig. 2A and B, lane 3). Upon
analyzing the soluble fraction and inclusion bodies, the
recombinant protein N60-GST was found to circulate in the soluble
fraction (Fig. 2A, lane 5), while
the majority of the recombinant protein C45-GST was present in the
inclusion bodies (Fig. 2B, lane 6).
Since the recombinant protein C45-GST was present in the insoluble
fraction, the N60-GST and C45-GST proteins were purified by
electroelution, obtaining at least 80 mg/100 ml of each recombinant
protein subsequent to desalting using the Sephadex G-25 column
(Fig. 2A and B, lane 7).
Production of polyclonal antisera
against recombinant VP22 proteins
The antisera against VP22 N-terminal peptide
(labeled as anti-N60) and VP22 C-terminal peptide (labeled as
anti-C45) were collected from mice following three antigen
injections. ELISA and western blot analysis were used to validate
the reactivity and specificity of the antisera, respectively. The
antisera reacted at different dilutions (anti-N60 dilutions, 1:200
to 1:16,000; and anti-C45 dilutions, 1:200 to 1:32,000) with an
equal amount of the corresponding recombinant proteins (data not
shown). No positive signal was detected from the pre-immunized
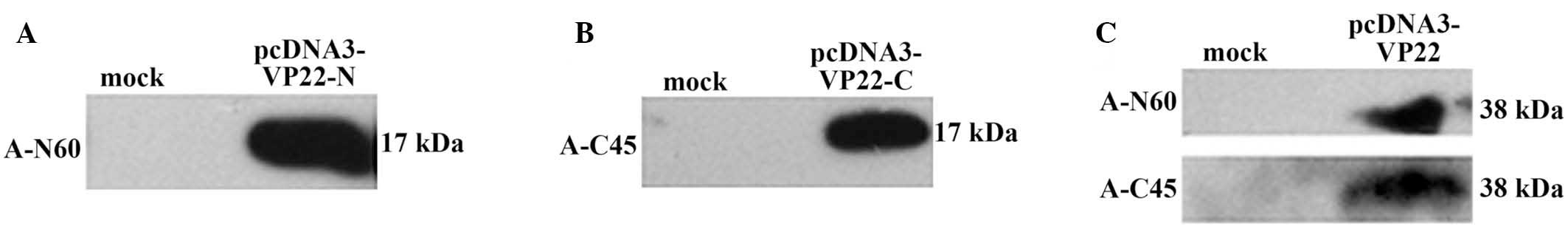
mouse serum, which acted as a negative control. The results
indicated that anti-N60 and anti-C45 were able to specifically
detect their respective truncated VP22 proteins (Fig. 3A and B) and full-length VP22 proteins
(Fig. 3C). In addition, the antisera
did not show any non-specific reaction against the
pcDNA3-transfected cell lysates. The molecular weight of the
N-terminal 150 aa, the C-terminal 151 aa and the full-length VP22
protein were confirmed to be approximately 17 kDa, 17 kDa and 38
kDa, respectively.
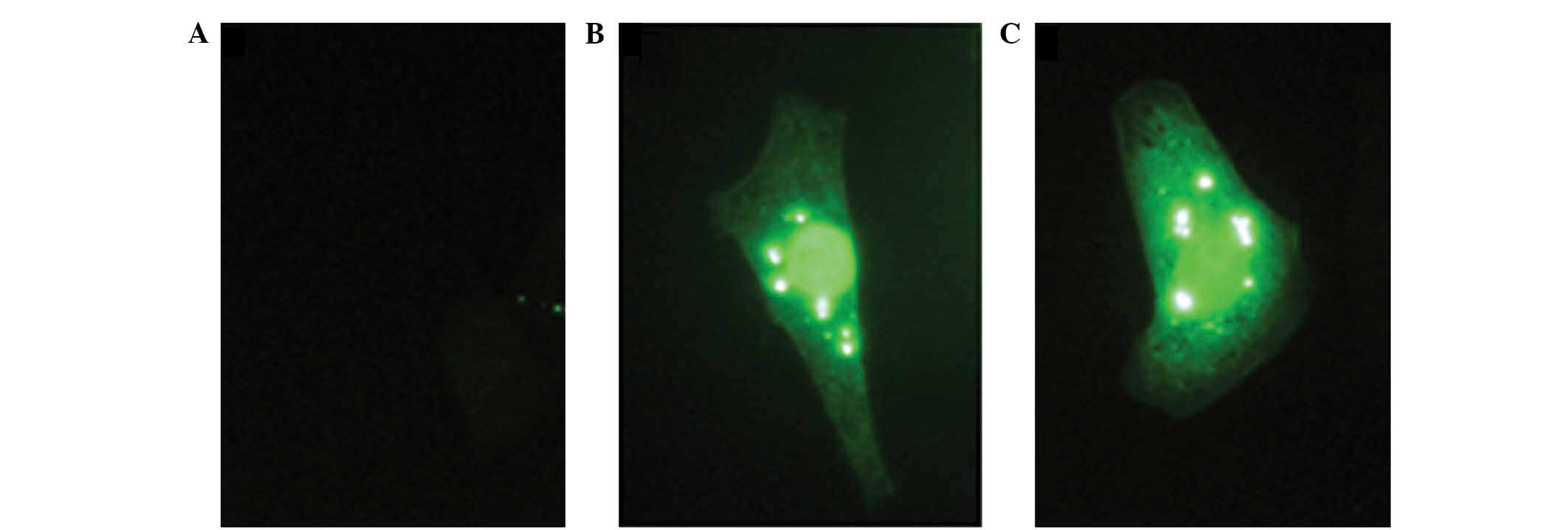
To validate the different uses of the polyclonal
antibody investigated in the present study, the subcellular
localization of VP22 protein in pcDNA3-VP22-transfected Vero cells
by immunofluorescence was examined. Pre-immunized mouse serum was
used as the negative control (Fig.
4A). The results indicated that anti-N60 (Fig. 4B) and anti-C45 (Fig. 4C) detected the full-length VP22,
while they showed medium cytosolic and predominant nuclear
localization of VP22 in the Vero cells. In conclusion, in the
present study, polyclonal antisera against the N- and C-terminus of
VP22 that recognizes the truncated and full-length VP22 protein
were developed. These antisera may serve as a useful tool for
further studies of VP22-fusion protein drugs, and facilitate the
development of these protein drugs in future studies.
Discussion
CPP-based intracellular delivery is one of the most
powerful techniques used for importing protein drugs into
eukaryotic cells (13,14). However, the lack of tools to track
VP22-fusion therapeutic molecules in cells may be a limitation in
the development of novel VP22-based treatments. The antibodies
generated against the C- and N-terminus of the VP22 protein may be
used to identify the cellular location of the VP22-fusion drugs.
The C-terminus of VP22 protein is essential for intercellular
transport (9,15), and the polyclonal C-terminal antibody
may be used to detect the position of C-terminal VP22-fusion
protein drugs in the cells. The truncated recombinant proteins were
cloned in frame with the N-terminal GST-tag of the pGEX-5X-1
vector.
Antibodies against different specific regions of the
VP22 proteins are increasingly used in the development of VP22
fusion protein drugs (7,16). In the present study, antisera against
the truncated VP22 peptides recognized their specific targets and a
full-length VP22 protein. These antisera can be used in various
molecular applications, including ELISA, western blot analysis and
immunofluorescence. Notably, the molecular weights of N60-GST,
C45-GST, N-terminal 150 aa and C-terminal 151 aa of VP22 were
consistent with the calculated theoretical values. The molecular
weight of the full-length VP22 protein was consistent with that of
the basic phosphorylated VP22 (17).
Furthermore, the cytosolic and nuclear localization of VP22 was
consistent with that reported previously (18), thus further validating the
antisera.
Currently, a limitation of large molecular drug
therapy (with genes or proteins) is the inability to deliver
sufficient amounts of active large molecular drugs to the target
cells (19). While secreted proteins
can overcome this limitation to a certain degree, this is
challenging for non-secreted proteins. In these cases, the
potentially therapeutic protein is only active in cells to which it
is initially delivered. It has been suggested that VP22 fusion
proteins may be able to provide a solution by increasing drug
distribution through intercellular delivery (8,20).
Theoretically, more cells would then reach a therapeutic
steady-state, leading to an overall enhancement of biological
effects in the target population. Further studies should
investigate the antitumor activity of VP22-fused antitumor protein
drugs and evaluate the immune response of VP22-fused antigens.
In conclusion, in the present study, polyclonal
antisera against VP22 were developed. These antisera will
facilitate the development of VP22-fusion protein drugs in future
studies.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81102288) and
Chongqing Science & Technology Commission (no.
cstc2014jcyjA10009).
References
|
1
|
Zahid M and Robbins PD: Cell-type specific
penetrating peptides: Therapeutic promises and challenges.
Molecules. 20:13055–13070. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gautam A, Singh H, Tyagi A, Chaudhary K,
Kumar R, Kapoor P and Raghava GP: CPPsite: A curated database of
cell penetrating peptides. Database (Oxford). 2012:bas0152012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Madani F, Lindberg S, Langel U, Futaki S
and Gräslund A: Mechanisms of cellular uptake of cell-penetrating
peptides. J Biophys. 2011:4147292011.PubMed/NCBI
|
|
4
|
Jiao CY, Delaroche D, Burlina F, Alves ID,
Chassaing G and Sagan S: Translocation and endocytosis for
cell-penetrating peptide internalization. J Biol Chem.
284:33957–33965. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elliott G and O'Hare P: Intercellular
trafficking and protein delivery by a herpesvirus structural
protein. Cell. 88:223–233. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanaka M, Kato A, Satoh Y, Ide T, Sagou K,
Kimura K, Hasegawa H and Kawaguchi Y: Herpes simplex virus 1 VP22
regulates translocation of multiple viral and cellular proteins and
promotes neurovirulence. J Virol. 86:5264–5277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zavaglia D, Favrot MC, Eymin B, Tenaud C
and Coll JL: Intercellular trafficking and enhanced in vivo
antitumour activity of a non-virally delivered P27-VP22 fusion
protein. Gene Ther. 10:314–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin G, Zhou Y, Chai Q, Zhu G, Xu F and Liu
F: VP22 and cytosine deaminase fusion gene modified
tissue-engineered neural stem cells for glioma therapy. J Cancer
Res Clin Oncol. 139:475–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nishikawa M, Otsuki T, Ota A, Guan X,
Takemoto S, Takahashi Y and Takakura Y: Induction of tumor-specific
immune response by gene transfer of Hsp70-cell-penetrating peptide
fusion protein to tumors in mice. Mol Ther. 18:421–428. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bian J, Popovic ZB, Benejam C, Kiedrowski
M, Rodriguez LL and Penn MS: Effect of cell-based intercellular
delivery of transcription factor GATA4 on ischemic cardiomyopathy.
Circ Res. 100:1626–1633. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu X, Liu L, Wu L, Wang L, Dong C, Li W
and Li Q: Herpes simplex virus type 1 tegument protein VP22 is
capable of modulating the transcription of viral TK and gC genes
via interaction with viral ICP0. Biochimie. 92:1024–1030. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Feng S, Huang Y, Qiao M, Zhang B
and Xu H: Prokaryotic expression, purification and polyclonal
antibody production of a hydrophobin from Grifola frondosa. Acta
Biochim Biophys Sin (Shanghai). 42:388–395. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bolhassani A: Potential efficacy of
cell-penetrating peptides for nucleic acid and drug delivery in
cancer. Biochim Biophys Acta. 1816:232–246. 2011.PubMed/NCBI
|
|
14
|
Sawant R and Torchilin V: Intracellular
transduction using cell-penetrating peptides. Mol Biosyst.
6:628–640. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aints A, Güven H, Gahrton G, Smith CI and
Dilber MS: Mapping of herpes simplex virus-1 VP22 functional
domains for inter-and subcellular protein targeting. Gene Ther.
8:1051–1056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perkins SD, Flick-Smith HC, Garmory HS,
Essex-Lopresti AE, Stevenson FK and Phillpotts RJ: Evaluation of
the VP22 protein for enhancement of a DNA vaccine against anthrax.
Genet Vaccines. 3:32005. View Article : Google Scholar
|
|
17
|
Mouzakitis G, McLauchlan J, Barreca C,
Kueltzo L and O'Hare P: Characterization of VP22 in Herpes simplex
virus-infected cells. J Virol. 79:12185–12198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yedowitz JC, Kotsakis A, Schlegel EF and
Blaho JA: Nuclear localizations of the herpes simplex virus type 1
tegument proteins VP13/14, vhs and VP16 precedeVP22-dependent
microtubule reorganization and VP22 nuclear import. J Virol.
79:4730–4743. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi NQ, Qi XR, Xiang B and Zhang Y: A
survey on ‘Trojan Horse’ peptides: Opportunities, issues and
controlled entry to ‘Troy’. J Control Release. 194:53–70. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu X, Xu Z, Lei J, Li T and Wang Y: VP22
mediates intercellular trafficking and enhances the in vitro
antitumor activity of PTEN. Mol Med Rep. 12:1286–1290.
2015.PubMed/NCBI
|