Introduction
Peripheral nerve injury is a complication that is
commonly observed following orthopedic surgery or trauma (1–3).
Peripheral nerve ischemia and reperfusion (I/R), which may be the
cause of nerve injury, may occur following incision for
pressure-relief in extremity intracompartment syndrome, during
compression for repair of vascular rupture or from nerve trunk
oppression (4). Numerous interacting
mechanisms may contribute to axonal degeneration following I/R,
including immune cell activation/infiltration, calcium
dysregulation, and free radical generation (5).
In the central nervous system (CNS), glutamate
release and the concomitant overstimulation of synaptic glutamate
receptors, particularly the N-methyl-D-aspartate (NMDA) subtype, is
a critical early event in I/R injury (6). The involvement of glutamate receptors
in I/R injury has been extensively studied in the CNS (5); however, peripheral nerves also express
glutamate receptors, which may contribute to axonal injury and
nerve dysfunction, including initiation of neuropathic pain
(7–13). Oxidative stress is a hallmark of
peripheral nervous system (PNS) axon I/R injury, and occurs due to
the generation of free-radicals (4,14–17).
Free-radical scavengers have previously been shown to mitigate I/R
injury in the PNS (18). Activation
of nitric oxide (NO) synthase (NOS) may induce oxidative damage
during I/R (19–22); NO is highly reactive and may form
toxic reactive nitrogen species during reperfusion (23,24).
Conversely, as a major vascular relaxation factor, NO may also
protect against ischemic injury (25).
The pro-inflammatory cytokine tumor necrosis factor
(TNF)-α has previously been shown to be upregulated in ischemic
peripheral nerves (26,27). Conversely, downregulation of TNF-α
expression levels appeared to protect peripheral nerve integrity
and mitigate neurological sequela (28). TNF-α activity is influenced by the
TNF-α converting enzyme (TACE) (29). Therefore, TACE has been considered a
potential therapeutic target for CNS inflammatory disorders
(29); however, there is limited
information regarding its function in the PNS. In a previous study,
stress-induced increases in TACE activity, and increased expression
levels of TNF-α, were blocked by the noncompetitive NMDA receptor
antagonist MK-801 (30). This was
consistent with the results of previous studies, which demonstrated
neuroprotective effects for MK-801 in various models of strokes
(31,32).
The present study hypothesized that MK-801 may
protect peripheral nerves against I/R injury by inhibiting the
activation of TNF-α by TACE, and thereby suppressing
inflammation-mediated damage, and by inhibiting the activation of
iNOS, and thereby suppressing oxidative damage. Therefore, the
present study aimed to investigate the effects of MK-801 on the
expression levels of TNF-α, TACE and iNOS, and the peripheral nerve
histology, in a rat model of I/R injury.
Materials and methods
Animals
A total of 104 male Sprague-Dawley rats (age, 8–9
weeks; weight, 250–300 g) were supplied by the Animal Laboratory of
the Fujian Medical University (Fuzhou, China). The rats were
maintained under a 12-h light/dark cycle at 20–25°C and 50±5%
humidity, with ad libitum access to food and water. The rats
were acclimatized for 1 week prior to the experiments. All
procedures and animal experiments were approved by the Animal Care
and Use Committee of the Fujian Medical University, and were
conducted in accordance with all state regulations.
Rat model of sciatic nerve I/R
injury
The rat model of SN I/R injury was established as
described in a previous study (33).
Briefly, the rats were fasted for 12 h, with ad libitum
access to water, prior to experiments. Subsequently, the rats were
anesthetized by intraperitoneal injection with 3% pentobarbital
sodium (30 mg/kg; Maixin Biological Technology Development Co.,
Fuzhou, China) and placed onto the operating board in the supine
position. The groin on the right side was depilated using a knife
and disinfected with iodine complex, after which a bevel cut was
made in the right lower quadrant. In order to induce lower limb
ischemia, the right arteria iliaca communis, femoral artery and
arteria circumflexa iliaca superficialis were clamped for 5 h with
micro-artery forceps using an extraperitoneal approach.
Animal grouping
The rats were randomly divided into the following
groups: i) Sham group (n=8); ii) I/R group (n=48); and iii)
I/R+MK-801 group (n=48). The rats in the sham group were subjected
to anesthesia, skin preparation involving depilation and
disinfection with iodine, and bevel cutting into the right groin;
however, the arteries were not clamped. Blood samples (8 ml) were
obtained from all rats and the right SN were collected 24 h after
stitching of the wound.
The rats in the I/R group underwent I/R and were
injected intraperitoneally with 0.5 ml normal saline at 15 min
prior to reperfusion. The rats in the I/R+MK-801 group underwent
I/R and were intraperitoneally injected with 0.5 ml MK-801 (1
mg/kg; Sigma-Aldrich, St. Louis, MO, USA) at 15 min prior to
reperfusion. The rats in the I/R and I/R+MK-801 groups were divided
into six subgroups (n=8 per subgroup), depending on the time at
which they were sacrificed following reperfusion (0, 6, 12, 24, 72
h, or 7 days). Rats in the same subgroup did not vary in body
weight by >10 g. Blood samples were collected from the jugular
vein immediately prior to sacrifice. The rats were sacrificed by
acute blood loss following anesthetization with 10 mg/kg xylazine
hydrochloride (Maixin Biological Technology Development Co.) and 70
mg/kg ketamine (Maixin Biological Technology Development Co.).
Following sacrifice, a 5-cm section of the ipsilateral SN, ending 2
cm above the right knee joint, was removed.
Detection of iNOS activity in SN
tissues
A 0.5-g sample of SN was rinsed with ice-cold normal
saline for removal of blood, dried with filter paper, and
homogenized in ice-cold normal saline (dilution, 1:9) containing
0.86% sodium chloride. The concentration of iNOS was detected using
a Nitric Oxide Synthase typed assay kit (A014-1; Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) and a type 721
spectrophotometer (Shanghai Precision & Scientific Instrument
Co., Ltd., Shanghai, China). The protein concentration was
quantified using a Bicinchoninic Acid Protein Assay kit (Beyotime
Institute of Biotechnology, Nantong, China), according to the
manufacturer's instructions. Activity of iNOS is expressed per mg
of proteins.
Plasma NO levels
The plasma concentration of NO was determined using
a commercial NO Assay kit (A012; Nanjing Jiancheng Bioengineering
Institute), according to the manufacturer's protocol. Absorbance
was measured at 550 nm using the type 721 spectrophotometer
(Shanghai Precise Scientific Instrument Co., Ltd.).
Plasma malondialdehyde (MDA)
levels
The plasma concentration of MDA was detected in 0.1
ml plasma samples using the MDA Assay kit (A003-1; Nanjing
Jiancheng Bioengineering Institute), according to the
manufacturer's protocol. Absorbance was measured at 721 nm using a
type 721 spectrophotometer (Shanghai Precise Scientific Instrument
Co., Ltd.).
Hematoxylin & eosin (H&E)
staining
SNs were collected, fixed with 10% formaldehyde and
embedded in paraffin. Slices (4 µm) were prepared and heated at
60°C for 6 h to dry. Following removal of paraffin, the slices were
stained with H&E (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and visualized under an inverted phase contrast microscope
(Olympus Corporation, Tokyo, Japan).
Immunohistochemical analysis
Protein expression levels of TNF-α in the SN were
measured using the Elivision TM Plus Polymer HRP (Mouse/Rabbit)
Immunohistochemistry kit (Maixin Biological Technology Development
Co.) based on streptavidin-biotin-peroxidase visualization (Wuhan
Boster Bio-Engineering Co., Ltd., Wuhan, China). Tissue slices were
incubated with rabbit anti-mouse TNF-α polyclonal antibody (1:100;
BA14901; Wuhan Boster Bio-Engineering Co., Ltd.) at 4°C overnight.
After washing in phosphate-buffered saline (Maixin Biological
Technology Development Co.), labeled slices were incubated with
biotin-conjugated goat anti-mouse immunoglobulin G polyclonal
antibody (1:10,000; BA1001; Wuhan Boster Bio-Engineering Co., Ltd.)
at 25°C for 20 min, followed by incubation with a
streptavidin-biotin-peroxidase complex for 20 min at 25°C. Staining
was visualized by incubating the tissue sections with 99%
diaminobenzidine solution (Maixin Biological Technology Development
Co.) at room temperature for 3–5 min, followed by counterstaining
with hematoxylin and mounting with fluorescent mounting medium
(DakoCytomation, Glostrup, Denmark). Immunostaining was
semi-quantified using the Image-Pro Plus software, version 5 (Media
Cybernetics, Inc., Rockville, MD, USA). Total cell numbers and the
fluorescence intensity of each cell were counted and quantified in
six separate fields (75–100 cells/field) for each of the
conditions. The relative fluorescence intensity was calculated by
dividing the total integrated optical density (IOD) by the total
number of cells in each field. Mean fluorescence intensity
measurements were. All sections were examined by a pathologist
blinded to the grouping.
Electron microscopy
SNs were fixed in a solution containing 25% glutaric
dialdehyde (Maixin Biological Technology Development Co.) and 1.5%
paraformaldehyde (Nanjing Sen Beijia biotechnology Co., Ltd.,
Nanjing, China). Following fixation with 1% osmium tetroxide
(Maixin Biological Technology Development Co.), and dehydration
with ethanol and acetone, the SNs were embedded in epoxy resin 618
(Maixin Biological Technology Development Co.). Ultrathin sections
were prepared with a LKB-5 Ultramicrotome (GE Healthcare Life
Sciences, Uppsala, Sweden), double-stained with uranyl acetate and
calcium citrate (both Maixin Biological Technology Development
Co.), and examined using an HU-12A Transmission Electron Microscope
(Hitachi, Ltd., Tokyo, Japan).
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from SN tissues (0.1 mg)
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). RNA purity was determined by measuring the
absorbance ratio at 260 and 280 nm (A260/A280) using a NanoVue UV
spectrophotometer (GE Healthcare Life Sciences). cDNA was
synthesized using the PrimeScript First Strand cDNA Synthesis kit
(Takara Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's protocol. PCR amplification was performed in a
volume of 50 µl containing 200 ng cDNA, 25 pmol/l of each primer,
0.25 mmol/l deoxyribonucleotide triphosphates, 5 µl 10X buffer,
21.5 mmol/l MgCl and 2.0 U Taq HS polymerase (Takara
Biotechnology Co., Ltd.). Primers for the PCR were as follows:
TNF-α forward, 5′-CAAACCACCAAGCAGAGGAGC-3′ and reverse,
5′-CAAAGTGAGCTTGCCCGGACT-3′; TACE forward,
5′-CACTTTGGTGCCTTTCGTCC-3′ and reverse, 5′-AGCTCGCCTCTTCGCTCGAC-3′;
and β-actin forward, 5′-ATCCGTAAAGACCTCTATGC-3′ and reverse,
5′-AACGCAGCTCAGTAACAGTC-3′ (BIO Asia Biotechnology Co., Ltd.,
Shanghai, China). PCR cycling conditions were as follows: An
initial denaturation step at 94°C for 3 min, followed by 35 cycles
of 94°C for 30 sec, 52/48°C for 30 sec (TNF-α/TACE), and 72°C for
60 sec. The PCR products were subjected to agarose gel
electrophoresis and the abundance of each mRNA was normalized
against β-actin using the Gel Doc 1000 UV Gel Imaging system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
SPSS software, version 10.0 (SPSS, Inc., Chicago,
IL, USA) was used to conduct statistical analyses. Data are
presented as the mean ± standard deviation. Homogeneity of variance
was evaluated using the Levene's test. In the case of homogeneity,
one-way analysis of variance was conducted, followed by pair-wise
between- and within-group comparisons using least significant
difference (LSD) tests. In the case of heterogeneity, the
Games-Howell test was used for analysis. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effects of MK-801 pretreatment on
I/R-induced iNOS activity in the SN
The SNs isolated from the sham-operated group
exhibited low levels of iNOS activity (Table I). In the I/R group, the activity of
iNOS was not significantly different to the sham group immediately
following reperfusion (0 h time point; P>0.05); however, it
gradually increased thereafter (P<0.05 at ≥6 h), and peaked at
24 h following reperfusion (P<0.01; Table I). The activity of iNOS was decreased
in the I/R group at >24 h post-reperfusion; however, it remained
significantly different to the sham group at 7 days
post-reperfusion (P<0.05; Table
I). In the I/R+MK-801 group, the activity of iNOS followed the
same temporal course as the I/R group; however, the activity was
markedly lower at each time point, and the differences were
significant at 12, 24, 72 h, and 7 days post-reperfusion, as
compared with the I/R group (all P<0.05).
 | Table I.Effects of MK-801 on siatic nerve
iNOS activity, and plasma NO and MDA levels following I/R
injury. |
Table I.
Effects of MK-801 on siatic nerve
iNOS activity, and plasma NO and MDA levels following I/R
injury.
|
| Time
post-reperfusion |
|---|
|
|
|
|---|
| Marker | 0 h | 6 h | 12 h | 24 h | 72 h | 7 days |
|---|
| iNOS (U/mg) |
|
|
Sham |
0.23±0.11 |
0.25±0.08 |
0.30±0.15 |
0.27±0.10 |
0.23±0.07 |
0.19±0.05 |
|
I/R |
0.29±0.10 |
0.62±0.13a |
1.68±0.18b |
2.45±0.25b |
1.80±0.21b |
0.68±0.25b |
|
I/R+MK-801 |
0.26±0.13 |
0.47±0.15a |
1.25±0.10b,c |
1.65±0.23b,d |
1.36±0.19b,c |
0.49±0.15a,c |
| NO (µmol/l) |
|
|
Sham |
38.35±5.12 |
43.28±6.17 |
50.63±8.25 |
48.43±9.52 |
42.83±5.92 |
35.74±6.29 |
|
I/R |
53.64±3.51a |
75.16±2.65b |
127.85±3.18b |
175.29±4.15b |
97.74±2.18b |
75.91±1.76b |
|
I/R+MK-801 |
48.03±4.13a |
60.15±1.77b,c |
84.78±3.78b,d |
107.25±3.62b,d |
63.65±1.47b,c |
55.28±2.76a,c |
| MDA (µmol/l) |
|
|
Sham |
4.52±0.62 |
4.81±1.18 |
6.12±1.30 |
5.34±1.11 |
4.82±0.96 |
4.23±0.45 |
|
I/R |
6.30±0.73 |
9.73±1.21a |
12.54±1.46b |
16.23±1.92b |
10.63±1.23b |
6.56±0.5a |
|
I/R+MK-801 |
5.95±0.65 |
7.22±1.05a,c |
9.08±1.20b,c |
11.83±1.64b,d |
7.95±0.81a,c |
5.14±0.52 |
Effects of MK-801 pretreatment on
I/R-induced increases in plasma NO and MDA levels
As compared with the sham group, the I/R group
exhibited elevated plasma levels of NO immediately following
reperfusion, which peaked at 24 h post-reperfusion, and remained
significantly elevated at 7 days post-reperfusion (P<0.05;
Table I). The mean plasma levels of
NO in the I/R+MK-801 group followed the same general trend;
however, the plasma levels of NO were always lower in the
I/R+MK-801 group, as compared with the I/R group, and were
significantly lower at 6, 12, 24, 72 h, and 7 days post-reperfusion
(P<0.05; Table I).
Plasma concentrations of MDA were significantly
elevated in the I/R group at all time points, as compared with the
sham group (P<0.05; Table I). In
addition, the I/R+MK-801 group exhibited lower mean plasma MDA
levels at all time points, as compared with the I/R group, and were
significantly different at 6, 12, 24 and 72 h post-reperfusion (all
P<0.05; Table I).
A temporal correlation between iNOS activity and
levels of NO suggested that iNOS activity is the primary source of
plasma NO, and lower plasma levels of MDA and NO in the I/R+MK-801
group, as compared with the I/R group, indicated that MK-801 may
reduce MDA levels by reducing the concentration of plasma NO.
Effects of MK-801 on I/R-induced
histological changes in the SN
SNs from the sham group exhibited no significant
histological alterations after 5 h of ischemia, with only moderate
swelling of endothelial cells and edema around the blood vessels
(Fig. 1A). At 6 h post-reperfusion,
neutrophils were adhered to the blood vessel walls and had
infiltrated the surrounding endoneurium and epineurium (Fig. 1B). At 12 h post-reperfusion, further
neutrophil adherence and infiltration was observed. In addition,
the myelin sheathes were vesicular in appearance, and degranulation
of neutrophils was detected. At 24 h post-reperfusion, the leakage
of neutrophils out of blood vessels and infiltration into the SN
reached a peak, and this was accompanied by detection of
neutrophils around myelinated fibers, adherence of monocytes to
vessels surrounding the endoneurium, and obvious edema and swelling
of the endoneurium and myelin sheathes (Fig. 1C). At 72 h post-reperfusion, marked
infiltration of monocytes was observed, macrophages were detected
around Schwann cells and axon demyelination was observed. At 7 days
post-reperfusion, numerous infiltrated macrophages and monocytes
were detected, and this was accompanied by widespread demyelination
and edema of the myelin sheathes, endoneurium and endothelial cells
(Fig. 1D).
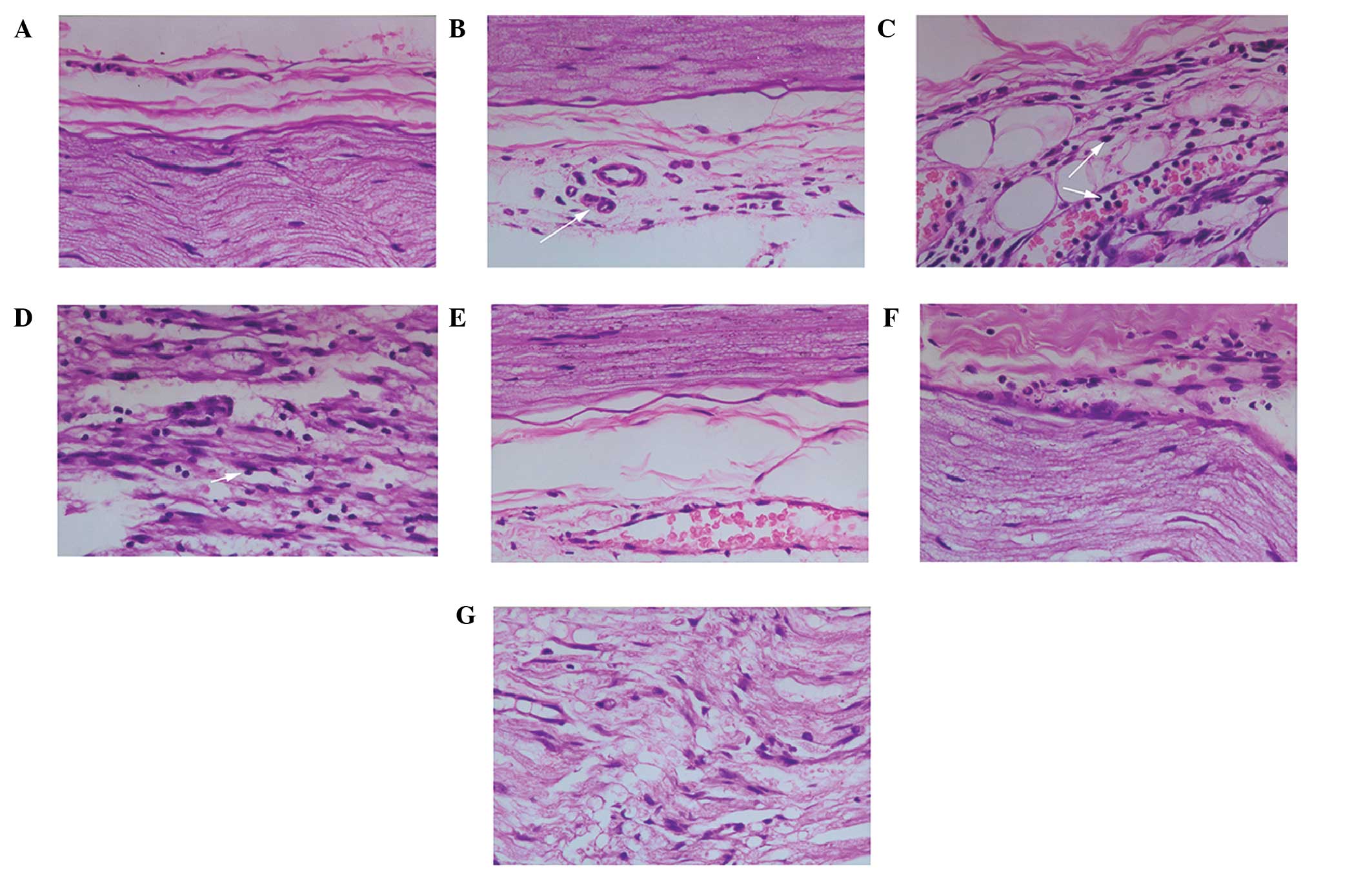 | Figure 1.Effects of MK-801 on histological
alterations in the rat sciatic nerve (SN) following
ischemia/reperfusion (I/R) injury. Histological changes were
determined using hematoxylin & eosin (H&E) staining
(magnification, ×400). (A) SN fibers and the outer membrane of
blood vessels from a sham-operated rat. (B) SN fibers and the outer
membrane of blood vessels from a rat in the I/R group at 6 h
post-reperfusion. Obvious edema around the epineurium, leakage and
infiltration of neutrophils around blood vessels, and swelling of
nerve fibers (arrow) were observed. (C) SN fibers and the outer
membrane of blood vessels from a rat in the I/R group at 24 h
post-reperfusion. Infiltration of neutrophils into the nerve fiber
bundle peaks, more severe fiber swelling, and partial demyelination
(white arrows) was observed. (D) SN fibers from a rat in the I/R
group at 7 days post-reperfusion. Large numbers of infiltrating
macrophages and monocytes were observed around the Schwann cells,
which was accompanied by extensive axonal demyelination. (E) SN
fibers and blood vessels surrounding the epineurium from a rat in
the I/R + MK-801 group at 6 h post-reperfusion. Pretreatment with
MK-801 resulted in milder edema around the epineurium, fewer
infiltrating neutrophils around blood vessels, and only minor
swelling of nerve fibers. (F) SN fibers and blood vessels around
the epineurium from a rat in the I/R + MK-801 group at 24 h
post-reperfusion. As compared with the I/R group rats at the same
post-reperfusion time point, there was less extensive axonal
swelling and edema of the endoneurium and myelin sheath, fewer
infiltrating neutrophils, and no observable demyelination. (G) SN
from a rat in the I/R + MK-801 group at 7 days post-reperfusion. As
compared with the I/R group rats at the same post-reperfusion time
point, swelling of myelin sheathes, infiltration of macrophages and
monocytes around Schwann cells, and demyelination were less
severe. |
No significant morphological alterations were
observed in the SNs from rats in the I/R+MK-801 group immediately
following reperfusion, and there was less edema, as compared with
the I/R group. At 6 and 12 h post-reperfusion, fewer adherent and
infiltrated neutrophils were observed, as compared with the I/R
group (Fig. 1E). At 24 h
post-reperfusion in the I/R+MK-801 group only mild inflammation was
observed, with comparatively less edema and swelling of the
endoneurium and myelin sheathes and no detectable demyelination
(Fig. 1F). At 72 h and 7 days
post-reperfusion, there remained fewer infiltrated monocytes
surrounding Schwann cells and less demyelination, as compared with
the I/R group (Fig. 1G).
Effects of MK-801 on I/R-induced
ultrastructural changes in the SN
SNs isolated from the sham, I/R and I/R+MK-801
groups were also examined by TEM. Consistent with the
histopathological observations, inflammatory cells were detected in
the blood vessels and surrounding the epineurium of the SNs from
the I/R group, which was accompanied by the adherence of
neutrophils to the blood vessel walls and the leakage of
neutrophils out of blood vessels. In addition, spaces between
endothelial cells were broadened, which indicated a loss of blood
vessel integrity (Fig. 2A). Certain
neutrophils were observed to be distant from the blood vessel
walls, and small numbers of lymphocytes were shown to surround
detached collagen fibers. Furthermore, deformed erythrocytes were
observed within various blood vessels, and signs of thrombosis and
platelet aggregation, with concomitant narrowing or even complete
blockage of blood vessels by fibrin and blood cells, was detected
(Fig. 2B). Small numbers of
infiltrating neutrophils and other inflammatory cells were detected
around nerve bundles, and fissures of various sizes due to detached
myelin were observed. In addition, swelling of various axons was
observed and irregularly shaped effusion cavities were shown to
have formed due to the separation of myelin sheathes from axons
(Fig. 2C). Certain myelin sheathes
showed signs of erosion, and various axons exhibited swollen
mitochondria and endoplasmic reticulum (ER). Swollen mitochondria
were also observed in the Schwann cells (Fig. 2D).
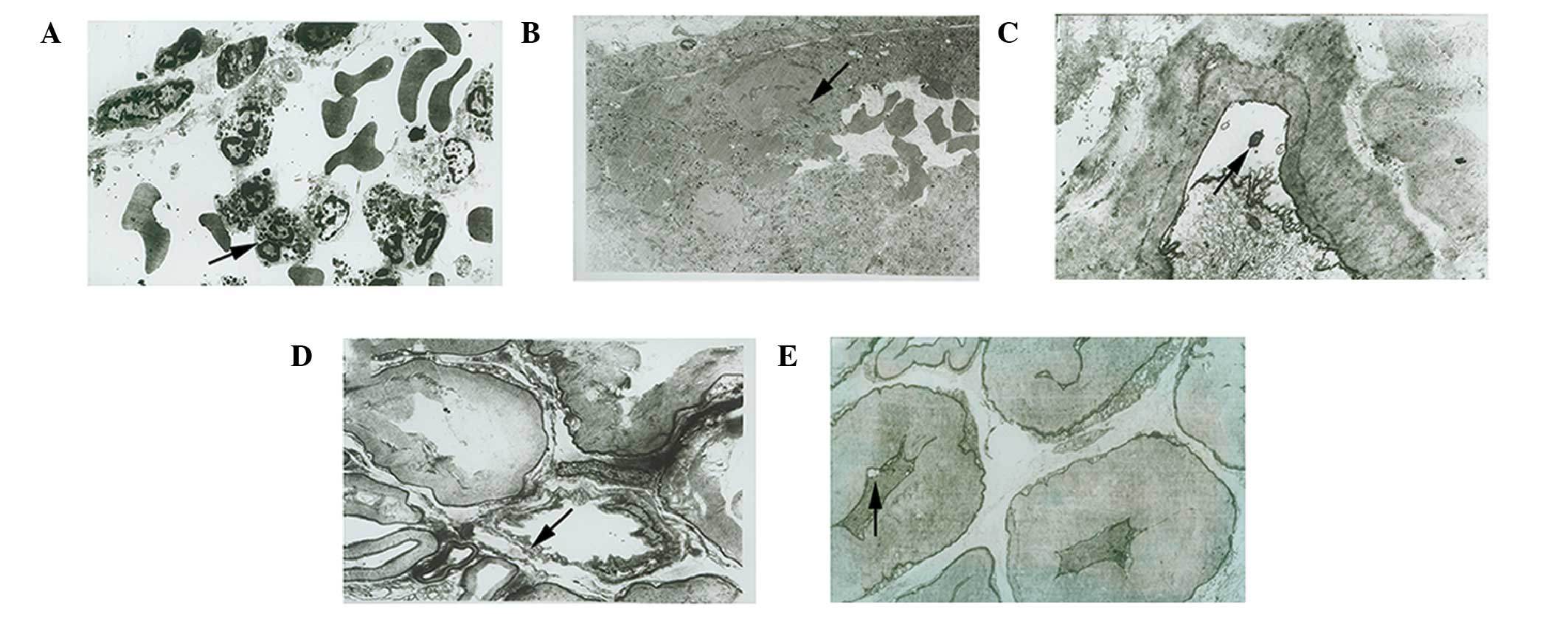 | Figure 2.Effects of MK-801 on ultrastructural
changes in the rat sciatic nerve (SN) following
ischemia/reperfusion (I/R) injury. The ultramicrostructure was
examined using transmission electron microscopy. (A) Blood vessels
surrounding the epineurium from a rat in the I/R group at 24 h
post-reperfusion (magnification, ×7,000). Neutrophils (black arrow)
were adhered to the blood vessel wall and were infiltrating the
nerve. (B) Blood vessels surrounding the epineurium from a rat in
the I/R group at 24 h post-reperfusion (magnification, ×7,000).
Thrombosis (black arrow), platelet aggregation and deformed
erythrocytes were detected. (C) Myelin sheathes in a SN from a rat
in the I/R group at 24 h post-reperfusion (magnification, ×10,000).
Fissures (black arrow) were detected between the axonal membrane
and myelin sheath. (D) Myelin sheathes in a SN from a rat in the
I/R group at 24 h post-reperfusion (magnification, ×7,000). Erosion
(black arrow) was detected. (E) Myelin sheathes in the SN from a
rat in the I/R + MK-801 group at 24 h post-reperfusion
(magnification, ×8,000). Myelin sheathes exhibited mild shrinkage
and occasional small fissures (black arrow) were observed between
the axonal membrane and myelin sheath. |
I/R injury-induced ultrastructural changes appeared
to be less severe in the SNs from the I/R+MK-801 group rats, which
exhibited fewer infiltrating inflammatory cells, swollen
mitochondria in axons and Schwann cells, and less severe ER
swelling. In addition, no thrombosis, platelet aggregation, or
clots were observed in the blood vessels of the SNs from the
I/R+MK-801 group. Furthermore, there were no signs of
demyelination, including fissures and effusion cavities, between
axons and myelin sheathes (Fig.
2E).
Effects of MK-801 on I/R-induced TNF-α
protein expression in the SN
SNs isolated from the sham group exhibited no
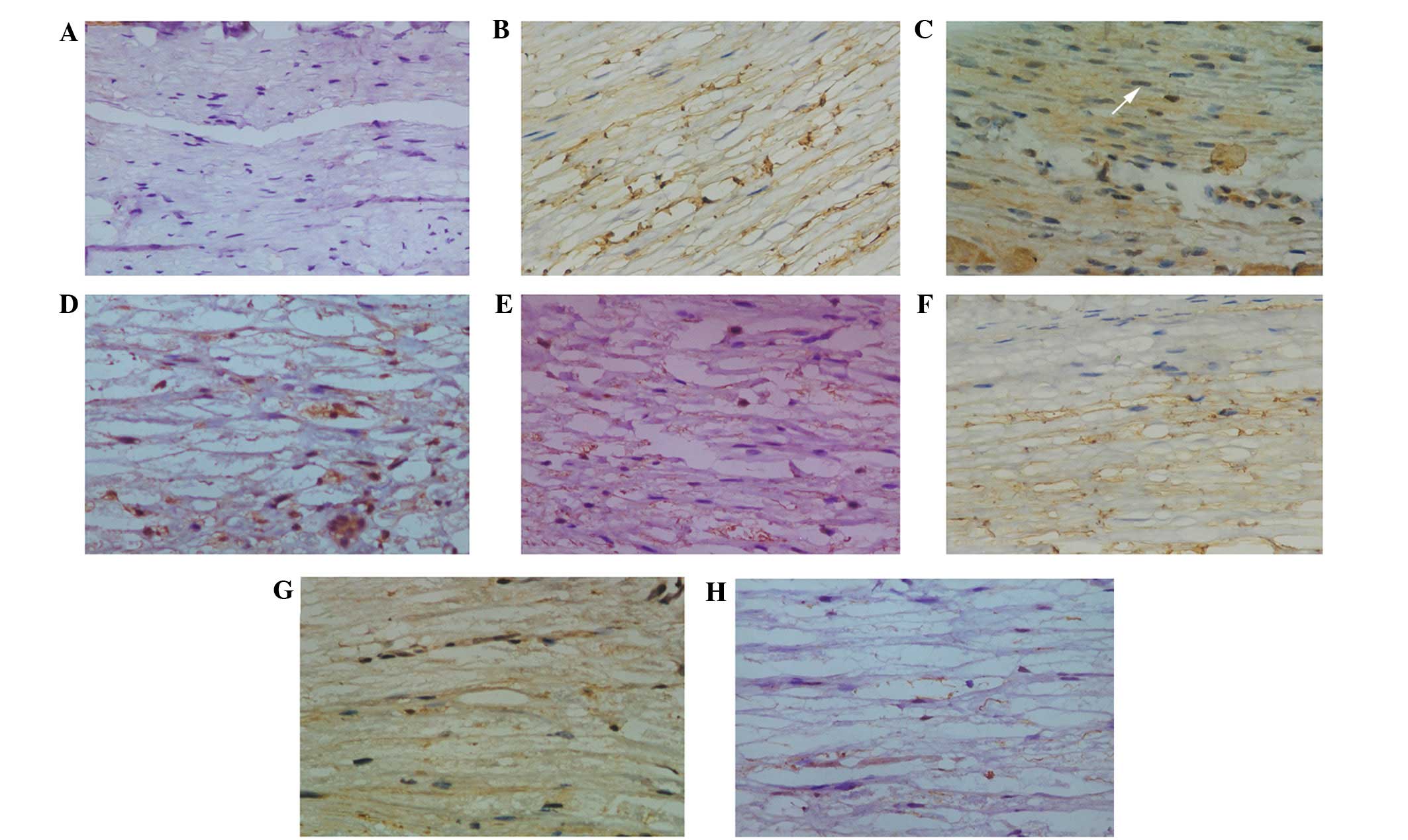
detectable expression of TNF-α in the Schwann cells (Fig. 3A). At the initiation of reperfusion
following 5-h ischemia, low protein expression levels of TNF-α were
detected in the Schwann cells. Concomitant with inflammatory cell
accumulation at the vessel walls and infiltration at 6–12 h
post-reperfusion, the protein expression levels of TNF-α increased
rapidly (Fig. 3B), and peaked at 24
h post-reperfusion (Fig. 3C). At 72
h post-reperfusion, the migration of neutrophils had ceased,
macrophages had infiltrated and were surrounding the Schwann cells,
demyelination was detected, and the protein expression levels of
TNF-α were gradually returned to baseline levels (Fig. 3D). At 7 days post-reperfusion,
widespread general demyelination was detected and the protein
expression levels of TNF-α had remained 2-fold higher, as compared
with baseline levels (Fig. 3E).
MK-801 pre-treatment markedly reduced the protein
expression levels of TNF-α during reperfusion (Fig. 3F–H), as demonstrated by a reduction
in the number of cells positive for TNF-α and the intensity of
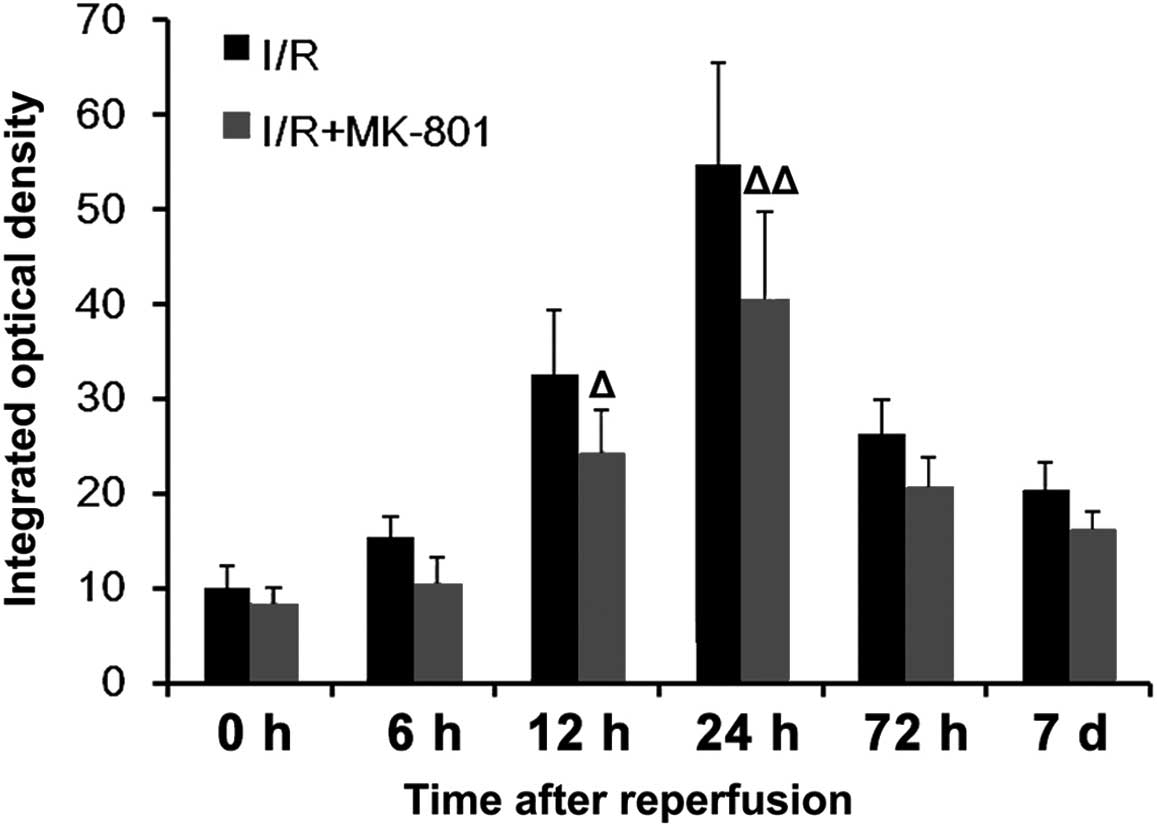
staining. IOD measurements demonstrated that the protein expression
levels TNF-α were significantly reduced at 12 and 24 h
post-reperfusion, as compared with the I/R group (Fig. 4).
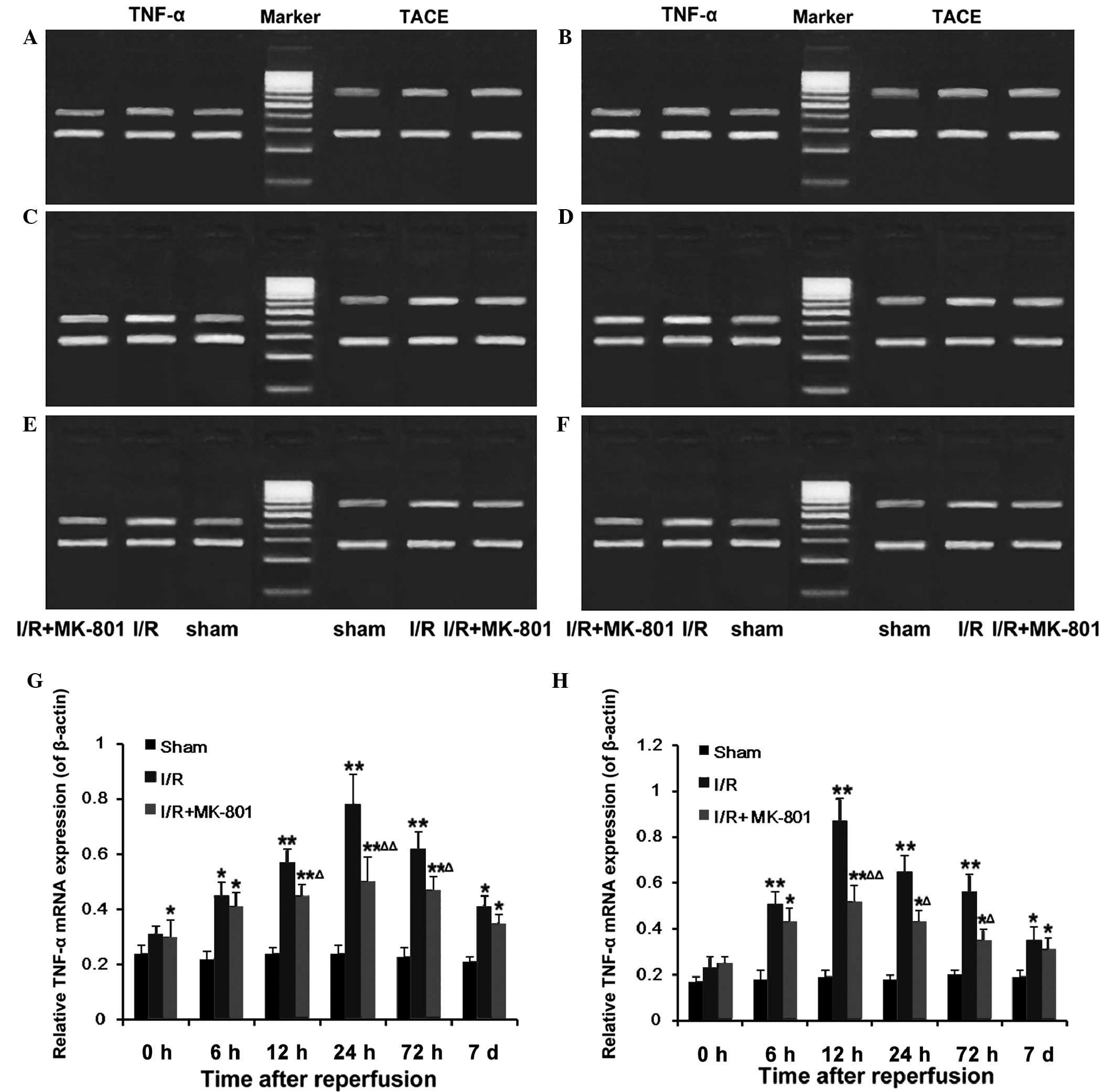
Effect of MK-801 on I/R-induced mRNA
expression of TNF-α and TACE in the SN
Following 5 h of ischemia, the mRNA expression
levels of TNF-α in the SNs from the I/R group rats were not
significantly different, as compared with the sham group
(P>0.05). However, reperfusion significantly increased the mRNA
expression levels of TNF-α, which peaked at 24 h post-reperfusion,
prior to decreasing to a level ~2-fold above the baseline at 7 days
post-reperfusion (Fig. 5A–G).
Similarly, the mRNA expression levels of TACE were increased during
reperfusion, and peaked at 12 h post-reperfusion, prior to
returning to the baseline at 7 days post-reperfusion (Fig. 5A–F and H). Pretreatment with MK-801
markedly reduced the mRNA expression levels of TNF-α and TACE
throughout the reperfusion period, with significant differences at
12–72 h post-reperfusion (P<0.05).
Discussion
Glutamate neurotoxicity (excitotoxicity) is a
seminal upstream event in the pathogenesis of I/R-induced neuronal
injury in the CNS (6). NMDAR is a
major contributor to this process, since it is the predominant
calcium-permeable glutamate receptor isoform and thus the major
link between the accumulation of extracellular glutamate from
dysregulated synaptic release and reverse transport (34–37), and
intracellular neurodegenerative pathways triggered by intracellular
calcium elevation, including proteolytic and free radical damage
(5,6,38). The
results of the present study suggested that NMDAR stimulation may
be a critical early trigger of I/R-induced neuronal injury in the
PNS. Notably, MK-801 pre-treatment was able to inhibit numerous
neurodegenerative downstream pathways in the SN, including edema,
cell swelling, mitochondrial damage, immune cell infiltration and
demyelination. Activation of TNF-α, TACE, and iNOS, which are
mediators of nerve inflammation and oxidative stress, may be a key
NMDAR-dependent intermediate step in this process, as rescue from
I/R-induced injury was associated with reduced iNOS, TNF-α, and
TACE expression levels, as well as decreased accumulation of MDA,
which is an indicator of oxidative stress.
The tissue microenvironment surrounding peripheral
nerves is distinct from the white matter of the CNS; thus major
differences in the underlying pathogenesis of I/R-induced nerve
injury in the CNS and the PNS may be expected. For example, the
source of the excitotoxic glutamate (synaptic vs. non-synaptic),
the role of the supporting cells (astrocytes vs. Schwann cells),
the immunoresponse, and the neuroprotective and cytotoxic
intermediates are distinct (39).
However, the rat SN exhibited a similar sequence of pathogenic
processes following I/R, as compared with the CNS, including early
edema and cell swelling, followed by delayed inflammation, axonal
degeneration and demyelination. Notably, these processes were
blocked or reduced in severity by MK-801 pre-treatment; thus
suggesting that early NMDAR activation may be involved in the
events leading to I/R injury (40,41).
In a previous study, peripheral application of
MK-801 was shown to prevent peripheral nerve damage (30), whereas activation of peripheral
glutamate receptors was shown to induce peripheral nerve damage or
dysfunction (30). For example, the
thermogenic flare induced by subdermal injection of the bee venom
toxin was associated with local extracellular glutamate
accumulation and was attenuated by co-injection with MK-801
(12); thus suggesting a role for
local NMDARs on sensory afferents as opposed to spinal NMDARs.
Similarly, peripheral glutamate released by damaged tissues and
reverse transport or glutamate influx from damaged capillaries, may
have induced SN damage during and following ischemia (37). Sodium influx upon NMDAR activation
may lead to axonal swelling by osmotic water movement, consistent
with the observed relief by MK-801 (42). Calcium influx associated with NMDAR
activation and ischemic-depolarization may initiate a chain of
biochemical events leading to axonal damage, including
calcium-dependent calcium release (43). At least within central axons, a form
of excitation-calcium coupling analogous to that in muscle cells
may exist, which induces calcium release from ryanodine-sensitive
stores (5,44). In addition, the ER is a major calcium
store (45), and the ultrastructural
investigations conducted in the present study detected signs of
axonal ER damage, possibly resulting in calcium egress. In turn,
dysregulated calcium may activate proteases that damage the axonal
cytoskeleton, resulting in disruption of axonal transport with
concomitant inhibition of retrograde growth factor signaling
(46). These same processes may also
be triggered at the proximal end of the nerve (closer to the spinal
cord), since glutamate within the dorsal root ganglia was enhanced
by SN injury, and cells within the dorsal root ganglia responded to
direct application of various glutamate receptor agonists,
including NMDA (13).
NO may relieve or exacerbate ischemic neural injury,
depending on context (47). As a
vasorelaxant, NO synthesized by neuronal NOS and iNOS may rescue
tissue by promoting reperfusion (48). Notably, ischemic preconditioning
involves iNOS induction (49).
Conversely, in previous studies, silencing iNOS exerted
neuroprotective effects against neurodegenerative diseases by
reducing microglial activation (50)
and/or oxidative stress (51). The
present study demonstrated a temporal correlation between iNOS
activation and lipid peroxidation during reperfusion; thus
suggesting that iNOS was a major source of free radicals in the
reperfused SN axons.
Comparable to NO, TNF-α may exert protective and
deleterious effects during ischemia (52,53),
with low levels mediating preconditioning, possibly by enhancing
the sensitivity of neurons to growth factors. However, TNF-α is a
central inflammatory mediator and higher concentrations have
previously been shown to be deleterious (26). In the present study, the reduction of
TNF-α levels by blocking NMDA receptors and reducing the expression
levels of TACE markedly suppressed the neuroinflammatory response,
leading to maintenance of the integrity of blood vessels, axonal
myelination, mitochondria, ER and Schwann cells during
reperfusion.
The present study included a number of limitations.
First, the clamping of the femoral artery induced I/R in the entire
limb; although only the SN was sampled and examined, the effects of
I/R on the entire limb may have affected the present results.
Second, the H&E sections and electron microscopy examinations
were analyzed subjectively. Third, the experimental approach did
not allow the determination of exact cause-to-effect relationships,
nor clarify the specific mechanisms underlying peripheral nerve I/R
injury. Furthermore, the lack of homology between rodent and human
anatomy, physiology and response to injury and inflammation may
limit the relevance of the results of the present study to humans.
Further studies are required in order to address these issues and
to improve the current understanding of peripheral nerve I/R
injury.
In conclusion, the present study demonstrated that
systemic injection of the NMDA receptor antagonist MK-801 was able
to protect the rat SN against I/R injury, including attenuating
immune cell infiltration and demyelination, possibly by inhibiting
the activation of TNF-α and reducing the expression levels of iNOS
in the SN.
Acknowledgements
The authors would like to thank the Fujian
Provincial Hospital Emergency Center of Fujian Province and the
Provincial Clinical Medical College of Fujian Medical University
for their assistance. The present study was supported by the
National Key Clinical Specialist Construction Programs of China
(grant no. 2006-1-45).
References
|
1
|
Hajek V, Dussart C, Klack F, Lamy A,
Martinez JY, Lainé P, Mazurier L, Guilloton L and Drouet A:
Neuropathic complications after 157 procedures of continuous
popliteal nerve block for hallux valgus surgery. A retrospective
study. Orthop Traumatol Surg Res. 98:327–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nassr A, Eck JC, Ponnappan RK, Zanoun RR,
Donaldson WF III and Kang JD: The incidence of C5 palsy after
multilevel cervical decompression procedures: A review of 750
consecutive cases. Spine (Phila Pa 1976). 37:174–178. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pavlovic D, Kocic G, Cvetkovic T, Simic D,
Basic J and Zivanovic D: Biomarkers of oxidative stress and
endothelial dysfunction after tourniquet release in children.
Physiol Res. 60(Suppl 1): S137–S145. 2011.PubMed/NCBI
|
|
4
|
Marin PC, Im MJ, Girotto JA, Borschel G
and Bickel KD: Effects of hydroxyethyl-starch-bound deferoxamine on
ischemia/reperfusion injury in chronic nerve compression. J
Reconstr Microsurg. 14:485–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stirling DP and Stys PK: Mechanisms of
axonal injury: Internodal nanocomplexes and calcium deregulation.
Trends Mol Med. 16:160–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Puyal J, Ginet V and Clarke PG: Multiple
interacting cell death mechanisms in the mediation of
excitotoxicity and ischemic brain damage: A challenge for
neuroprotection. Prog Neurobiol. 105:24–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou S, Bonasera L and Carlton SM:
Peripheral administration of NMDA, AMPA or KA results in pain
behaviors in rats. Neuroreport. 7:895–900. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ushida T, Tani T, Kawasaki M, Iwatsu O and
Yamamoto H: Peripheral administration of an N-methyl-D-aspartate
receptor antagonist (MK-801) changes dorsal horn neuronal responses
in rats. Neurosci Lett. 260:89–92. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ro JY: Contribution of peripheral NMDA
receptors in craniofacial muscle nociception and edema formation.
Brain Res. 979:78–84. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jang JH, Kim DW, Nam Sang T, Se Paik K and
Leem JW: Peripheral glutamate receptors contribute to mechanical
hyperalgesia in a neuropathic pain model of the rat. Neuroscience.
128:169–176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lam DK, Sessle BJ, Cairns BE and Hu JW:
Peripheral NMDA receptor modulation of jaw muscle electromyographic
activity induced by capsaicin injection into the temporomandibular
joint of rats. Brain Res. 1046:68–76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iwashita N, Nosaka S and Koyama N:
Involvement of peripheral NMDA receptor in melittin-induced
thermographic flare. Neurochem Res. 37:2222–2228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kung LH, Gong K, Adedoyin M, Ng J,
Bhargava A, Ohara PT and Jasmin L: Evidence for glutamate as a
neuroglial transmitter within sensory ganglia. PLoS One.
8:e683122013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He K, Nukada H, McMorran PD and Murphy MP:
Protein carbonyl formation and tyrosine nitration as markers of
oxidative damage during ischaemia-reperfusion injury to rat sciatic
nerve. Neuroscience. 94:909–916. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nagamatsu M, Schmelzer JD, Zollman PJ,
Smithson IL, Nickander KK and Low PA: Ischemic reperfusion causes
lipid peroxidation and fiber degeneration. Muscle Nerve. 19:37–47.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McCord JM: Oxygen-derived free radicals in
postischemic tissue injury. N Engl J Med. 312:159–163. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmelzer JD, Zochodne DW and Low PA:
Ischemic and reperfusion injury of rat peripheral nerve. Proc Natl
Acad Sci USA. 86:1639–1642. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iida H, Nagasaka T, Shindo K and Shiozawa
Z: Effect of the free radical scavenger edaravone on peripheral
nerve ischemia-reperfusion injury. Muscle Nerve. 40:582–588. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lefebvre RA: Nitric oxide in the
peripheral nervous system. Ann Med. 27:379–388. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cárdenas A, Moro MA, Hurtado O, Leza JC,
Lorenzo P, Castrillo A, Bodelón OG, Boscá L and Lizasoain I:
Implication of glutamate in the expression of inducible nitric
oxide synthase after oxygen and glucose deprivation in rat
forebrain slices. J Neurochem. 74:2041–2048. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu WH: Nitric oxide synthase in motor
neurons after axotomy. J Histochem Cytochem. 42:451–457. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qi WN, Yan ZQ, Whang PG, Zhou Q, Chen LE,
Seaber AV, Stamler JS and Urbaniak JR: Gene and protein expressions
of nitric oxide synthases in ischemia-reperfused peripheral nerve
of the rat. Am J Physiol Cell Physiol. 281:C849–C856.
2001.PubMed/NCBI
|
|
23
|
Chen XM, Chen HS, Xu MJ and Shen JG:
Targeting reactive nitrogen species: A promising therapeutic
strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol
Sin. 34:67–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shin SJ, Qi WN, Cai Y, Rizzo M, Goldner
RD, Nunley JA II and Chen LE: Inhibition of inducible nitric oxide
synthase promotes recovery of motor function in rats after sciatic
nerve ischemia and reperfusion. J Hand Surg Am. 30:826–835. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang RL, Zhang ZG and Chopp M: Targeting
nitric oxide in the subacute restorative treatment of ischemic
stroke. Expert Opin Investig Drugs. 22:843–851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wagner R and Myers RR: Endoneurial
injection of TNF-alpha produces neuropathic pain behaviors.
Neuroreport. 7:2897–2901. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niu YL, Guo Z and Zhou RH: Up-regulation
of TNF-alpha in neurons of dorsal root ganglia and spinal cord
during coronary artery occlusion in rats. Cytokine. 47:23–29. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stübgen JP: Tumor necrosis factor-alpha
antagonists and neuropathy. Muscle Nerve. 37:281–292. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lovering F and Zhang Y: Therapeutic
potential of TACE inhibitors in stroke. Curr Drug Targets CNS
Neurol Disord. 4:161–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kleinschnitz C, Brinkhoff J, Zelenka M,
Sommer C and Stoll G: The extent of cytokine induction in
peripheral nerve lesions depends on the mode of injury and NMDA
receptor signaling. J Neuroimmunol. 149:77–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ozyurt E, Graham DI, Woodruff GN and
McCulloch J: Protective effect of the glutamate antagonist, MK-801
in focal cerebral ischemia in the cat. J Cereb Blood Flow Metab.
8:138–143. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Corbett D, Evans S, Thomas C, Wang D and
Jonas RA: MK-801 reduced cerebral ischemic injury by inducing
hypothermia. Brain Res. 514:300–304. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nouri M, Rahimian R, Fakhfouri G, Rasouli
MR, Mohammadi-Rick S, Barzegar-Fallah A, Asadi-Amoli F and Dehpour
AR: Ipsilateral common iliac artery plus femoral artery clamping
for inducing sciatic nerve ischemia/reperfusion injury in rats: A
reliable and simple method. J Brachial Plex Peripher Nerve Inj.
3:272008.PubMed/NCBI
|
|
34
|
Hofmeijer J and van Putten MJ: Ischemic
cerebral damage: An appraisal of synaptic failure. Stroke.
43:607–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chao XD, Fei F and Fei Z: The role of
excitatory amino acid transporters in cerebral ischemia. Neurochem
Res. 35:1224–1230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Szydlowska K and Tymianski M: Calcium,
ischemia and excitotoxicity. Cell Calcium. 47:122–129. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grewer C, Gameiro A, Zhang Z, Tao Z,
Braams S and Rauen T: Glutamate forward and reverse transport: From
molecular mechanism to transporter-mediated release after ischemia.
IUBMB Life. 60:609–619. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma M: Role of calpains in the
injury-induced dysfunction and degeneration of the mammalian axon.
Neurobiol Dis. 60:61–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Richner M, Ulrichsen M, Elmegaard SL, Dieu
R, Pallesen LT and Vaegter CB: Peripheral nerve injury modulates
neurotrophin signaling in the peripheral and central nervous
system. Mol Neurobiol. 50:945–970. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mey J and Thanos S: Functional and
biochemical analysis of CNS-relevant neurotrophic activity in the
lesioned sciatic nerve of adult rats. J Hirnforsch. 37:25–50.
1996.PubMed/NCBI
|
|
41
|
Kim MA and Jeong KY: Chronological changes
of mechanical allodynia and spinal microglia activation by an
intrathecal injection of MK-801. Neuroreport. 24:585–589. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu XM: The Role of Intracellular Sodium in
the Regulation of NMDA-Receptor-Mediated Channel Activity and
Toxicity. Mol Neurobiol. 33:63–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xin WK, Kwan CL, Zhao XH, Xu J, Ellen RP,
McCulloch CA and Yu XM: A functional interaction of sodium and
calcium in the regulation of NMDA receptor activity by remote NMDA
receptors. J Neurosci. 25:139–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ouardouz M, Nikolaeva MA, Coderre E,
Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J and Stys
PK: Depolarization-induced Ca2+ release in ischemic spinal cord
white matter involves L-type Ca2+ channel activation of ryanodine
receptors. Neuron. 40:53–63. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sisalli MJ, Secondo A, Esposito A,
Valsecchi V, Savoia C, Di Renzo GF, Annunziato L and Scorziello A:
Endoplasmic reticulum refilling and mitochondrial calcium extrusion
promoted in neurons by NCX1 and NCX3 in ischemic preconditioning
are determinant for neuroprotection. Cell Death Differ.
21:1142–1149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morfini GA, Burns M, Binder LI, Kanaan NM,
LaPointe N, Bosco DA, Brown RH Jr, Brown H, Tiwari A, Hayward L, et
al: Axonal transport defects in neurodegenerative diseases. J
Neurosci. 29:12776–12786. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Brown GC: Nitric oxide and neuronal death.
Nitric Oxide. 23:153–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Phillips L, Toledo AH, Lopez-Neblina F,
Anaya-Prado R and Toledo-Pereyra LH: Nitric oxide mechanism of
protection in ischemia and reperfusion injury. J Invest Surg.
22:46–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Das M and Das DK: Molecular mechanism of
preconditioning. IUBMB Life. 60:199–203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li M, Dai FR, Du XP, Yang QD and Chen Y:
Neuroprotection by silencing iNOS expression in a 6-OHDA model of
Parkinson's disease. J Mol Neurosci. 48:225–233. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chavez-Valdez R, Martin LJ, Flock DL and
Northington FJ: Necrostatin-1 attenuates mitochondrial dysfunction
in neurons and astrocytes following neonatal hypoxia-ischemia.
Neuroscience. 219:192–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Taoufik E, Petit E, Divoux D, Tseveleki V,
Mengozzi M, Roberts ML, Valable S, Ghezzi P, Quackenbush J, Brines
M, et al: TNF receptor I sensitizes neurons to erythropoietin- and
VEGF-mediated neuroprotection after ischemic and excitotoxic
injury. Proc Natl Acad Sci USA. 105:6185–6190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Watters O and O'Connor JJ: A role for
tumor necrosis factor-α in ischemia and ischemic preconditioning. J
Neuroinflammation. 8:872011. View Article : Google Scholar : PubMed/NCBI
|