Introduction
Pulmonary arterial hypertension (PAH) is a fatal
condition characterized by increased pulmonary vascular resistance
and finally leading to right heart failure and mortality (1,2).
Endothelial injury, prolonged vasoconstriction and the
proliferation and migration of vascular smooth muscle cells
(VSMCs), are causes of increased pulmonary vascular resistance
(3). Multiple pharmacological
agents, such as vasodilators and anticoagulants, have been
developed for the treatment of PAH; however, the long-term
prognosis of patients with severe PAH remains poor (3). Therefore, there is an urgent
requirement for the development of more effective treatments for
PAH.
Rho-kinase (ROCK) is a member of the
serine/threonine kinase family that is an important downstream
effector of the small GTP-binding protein RhoA. The Rho/ROCK
pathway plays an important role in various fundamental cellular
functions, including contraction, motility, proliferation and
migration (4,5). There are two isoforms of ROCK, namely
ROCK1 (Rho-kinase β) and ROCK2 (Rho-kinase α) (6). ROCK1 and ROCK2 are highly homologous
with regard to amino acid sequence and kinase domains, sharing ~65%
homology in amino acid sequence and 92% homology in their kinase
domains (6). Although the two
isoforms are ubiquitously expressed in invertebrates and
vertebrates, ROCK1 is expressed mainly in circulating inflammatory
cells and ROCK2 is expressed in vascular cells (7,8).
Homozygous ROCK1-deficient mice show open eyelids at birth and
omphalocele, whereas homozygous ROCK2-deficient mice die
embryonically because of placental dysfunction, suggesting that
ROCK1 and ROCK2 mediate different functions in different types of
cells (9,10). To date, to the best of our knowledge,
whether ROCK is responsible for the growth of pulmonary arterial
endothelial cells (PAECs) has not yet been evaluated.
The present study was conducted to investigate the
effect of hypoxia on the proliferation of pulmonary arterial
endothelial cells (PAECs) and the role of ROCK2 in the underlying
mechanism. The activity and expression of ROCK2 were evaluated in
PAECs under hypoxic conditions, and the growth and proliferation of
PAECs were evaluated. In addition, the effects of hypoxia on the
expression of cyclin D and cyclin A, and the attenuating effects of
either a ROCK inhibitor or ROCK2 small interfering RNA (siRNA) were
tested. The results may reveal an important underlying mechanism of
PAEC overgrowth in the progression of PAH.
Materials and methods
Materials
The Y27632 ROCK inhibitor was purchased from Cayman
Chemical Co. (Ann Arbor, MI, USA). Rabbit anti-myosin phosphatase
target subunit 1 (anti-MYPT1; 2634) and anti-phospho (p)-MYPT1
(4563) polyclonal antibodies, and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (7074) and anti-mouse IgG
(7076) secondary antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Antibodies against rabbit
polyclonal ROCK2 (BA1766) and mouse monoclonal β-actin (BM0626)
were purchased from Boster Wuhan Biological Technology, Co., Ltd.
(Wuhan, China). 5-Bromo-2′-deoxyuridine (BrdU) proliferation assay
kit was from EMD Millipore (Billerica, MA, USA). All other reagents
were from common commercial sources.
Cell preparation and culture
PAECs were isolated from fresh bovine pulmonary
tissues as previously described (11). The bovine tissues were obtained from
a local slaughterhouse with all protocols reviewed and approved by
the Ethics Committee of Laboratory Animals at Jiamusi University
(Jiamusi, China). The identity was confirmed by typical endothelial
cell morphology and by positive anti-factor VIII staining. The
cells were cultured with Dulbecco's modified Eagle's medium (DMEM)
supplemented with 20% fetal bovine serum (FBS; both Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in a 37°C, 5% CO2
humidified incubator. Passages 2–5 were used for further
experimentation.
Small interfering RNA (siRNA) design
and transfections
PAECs were transfected with ROCK2 siRNA (siROCK2),
which was designed and synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). Non-targeted control siRNA (siNC) was used as a
negative control. The transfection protocol was that PAECs were
cultured until 30–50% confluence and then 1.5 µg siRNAs and 7.5 µl
X-treme Gene transfection reagent (Roche Diagnostics, Shanghai,
China) were diluted in serum-free Opti-MEM-1 medium (Thermo Fisher
Scientific, Inc.), respectively. The siRNAs and transfection
reagent were gently mixed together. After incubating at room
temperature for 20 min, the mixture was added directly onto the
cells. Following transfection, cells were quiesced in DMEM for 24 h
and used as required.
Cell treatment and groups
Firstly, cells were divided into three groups:
Normoxia (20% O2), hypoxia (5% O2) and
hypoxia plus Y27632 (5% O2 + 1 µM Y27632). Following
24-h treatment, protein and RNA were extracted from these cells.
Secondly, cells were also divided into three groups: Normoxia +
siControl, hypoxia + siControl and hypoxia + siROCK2. Following
24-treatment, protein and RNA were extracted from these cells.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay
PAECs were cultured at a density of
~1×104 per well in 96-well culture clusters, and then
the cells were treated with indicated reagents as groups. Dimethyl
sulfoxide (DMSO) and other agents at the indicated concentrations
were added for 24 h. Afterwards, the cells were incubated with 0.5%
MTT, which is a yellow mitochondrial dye, in sterile
phosphate-buffered saline for 4 h at 37°C. The reaction was
terminated by incubating the cells with DMSO for 10 min. The
absorbance at 540 nm was measured using an Epoch 2
spectrophotometer (BioTek China, Beijing, China). The amount of
blue formazan dye, formed from MTT, is proportional to the number
of surviving cells.
BrdU incorporation
PAECs were plated at 1×104 cells/well in
96-well plates, and then subjected to growth arrest for 24 h by
replacing the DMEM plus 1% FBS with DMEM prior to various
treatments. BrdU incorporation was measured using a BrdU
proliferation assay kit according to the manufacturer's protocol.
Briefly, the cells were labeled with 10 ng/ml BrdU during the
incubation, washed 3 times with cold wash buffer, fixed, air-dried
and incubated 1 h at room temperature with mouse anti-BrdU
monoclonal antibody (1:200). The antibody was aspirated, the cells
were washed three times and then incubated with HRP-conjugated goat
anti-mouse IgG (1:2,000) at room temperature for 30 min. The cells
were washed 3 times, and 100 µl substrate was added to each well
and incubated for 30 min in the dark. Thereafter, the absorbance
measured at dual-wave lengths between 450 and 540 nm was
determined.
Western blot analysis
Proteins from different experimental groups were
solubilized and extracted as previously reported (12). The protein concentrations were
determined by bicinchoninic acid protein assay (Pierce
Biotechnology, Inc., Rockford, IL, USA) with bovine serum albumin
(Thermo Fisher Scientific, Inc.) as a standard. Equal amounts of
protein (20 µg) from each sample were subjected to electrophoresis
on a 10% sodium dodecyl sulfate-polyacrylamide gel, and transferred
onto a nitrocellulose membrane (EMD Millipore). The blots were then
incubated in a blocking buffer (Tris 20 mM, pH 7.6, NaCl 150 mM,
and Tween 20 0.1%) containing 5% nonfat dry milk powder for 1 h at
room temperature. This was followed by incubation with anti-MYPT1
(1:1,000), anti-p-MYPT1 (1:1,000), anti-ROCK2 (1:250) and β-actin
(1:500) primary antibodies overnight at 4°C, and incubation with
HRP-conjugated goat anti-rabbit IgG and anti-mouse IgG secondary
antibodies (both 1:3,000) at room temperature and enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Inc.) the next
day. β-actin was used as the internal control in all
experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) and was reverse transcribed into
cDNA using a PrimeScript RT kit (Takara Biotechnology Co., Ltd.,
Dalian, China). Reverse transcription reaction mixture contained
added 2 µl 5X PrimeScript buffer, 0.5 µl PrimeScript RT enzyme mix
I, 0.5 µl oligo dT Primer (50 µM), 0.5 µl random hexamers (100 µM),
500 ng RNA and RNase-free dH2O to 10 µl, and was performed at 37°C
for 15 min and 85°C for 5 sec, followed by holding at 4°C. Applied
Biosystems (ABI) 7300 Fast Real-Time PCR system (Thermo Fisher
Scientific, Inc.) was used to perform qPCR experiments. Applied
Biosystems Primer Express 3.0 (Thermo Fisher Scientific, Inc.) was
used to design specific primers and BLAST analysis was used to
confirm the specificity of the primers. Each 20-µl reaction
contained 10 µM forward and reverse primers, 1X SYBR®
Premix Ex Taq™ II, 0.4 µl ROX reference dye (both Thermo Fisher
Scientific, Inc.) and 2 µl cDNA. The ABI 7300 system was programmed
with the PCR conditions: 95°C for 30 sec, 40 cycles of 95°C for 5
sec, and 60°C for 30 sec, and this was followed by routine melting
curve analysis. Primer sequences were as follows: ROCK2, forward
5′-CTAGGCCGGGCGAAGC-3′ and reverse 5′-CTCCAGCTTCCTCTGACGAC-3′;
cyclin D1, forward 5′-TCAAGTGTGACCCGGACTG-3′ and reverse
5′-AAGCCAGACCAGCTTCTTCC-3′; cyclin A, foward
5′-GCTGTGCGTTGCGGTTC-3′ and reverse 5′-GTGCGACTCCACTCTTCGAG-3′;
β-actin, forward 5′-AGGCCCCTCTGAACCCTAAG-3′ and reverse
5′-CCAGAGGCATACAGGGACAAC-3′. The target gene expression relative
quantitation (RQ) was calculated by the 2-ΔΔCq method
(13). The first step in the RQ
analysis was to normalize the target gene expression level to
β-actin (ΔCt) and the second step was to compare the difference in
normalized target gene expressions between different samples
(ΔΔCt). Each experiment was repeated 2–3 times in 3–4 samples.
Statistical analysis
Composite data are expressed as mean ± standard
error of the mean. Statistical analysis was performed with
Student's t-test or one-way analysis of variance followed by
Dunnett's test where appropriate. P<0.05 was considered to
indicate a statistically significant difference.
Results
Hypoxia increases the activity and
expression of ROCK2 in PAECs
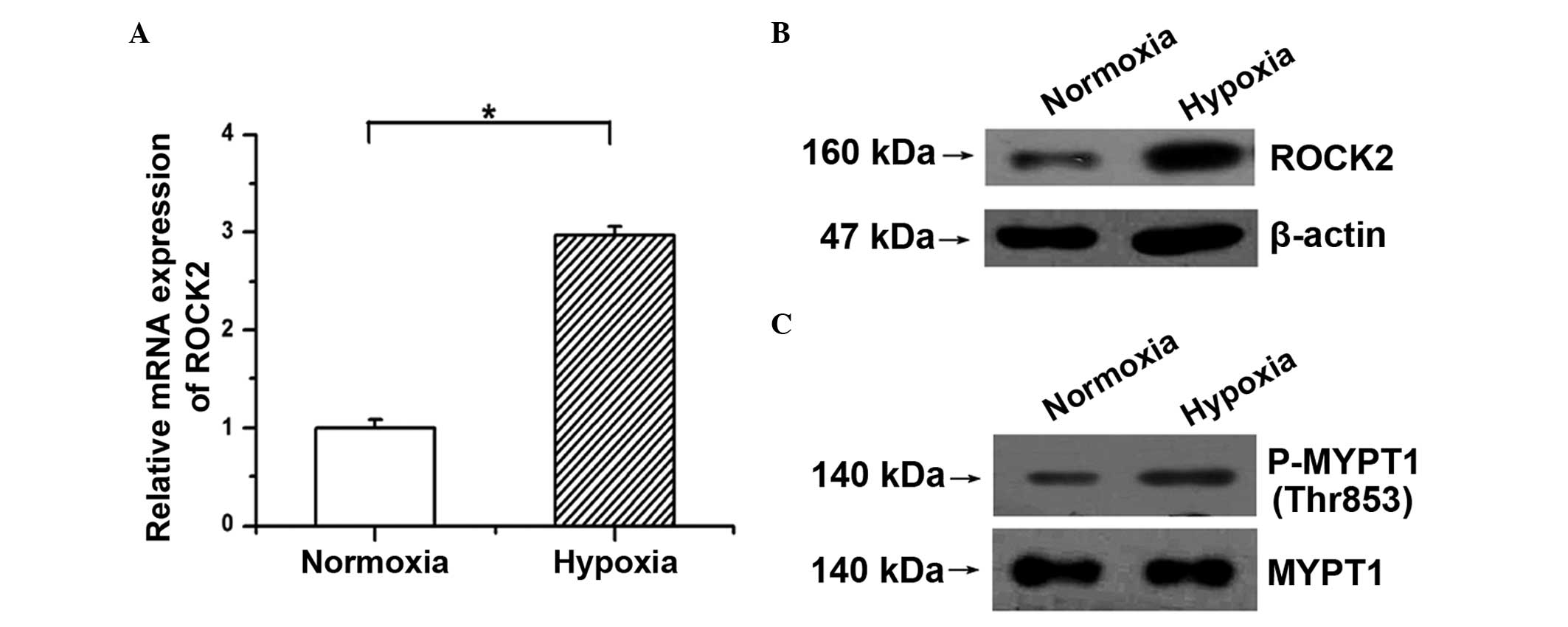
First, whether the ROCK2 pathway is activated under
hypoxic condition in PAECs was examined. As shown in Fig. 1A and B, hypoxia induced the mRNA and
protein expression of ROCK2 in PAECs. Moreover, as the
phosphorylation of MYPT1, a downstream target of ROCK, reflects
ROCK2 activity, the amounts of p-Thr853 MYPT1 and total MYPT1 were
determined. The western blotting results showed that the
phosphorylation of MYPT1 was significantly increased in PAECs
following stimulation with hypoxia compared with the normoxic group
(Fig. 1C). These results indicate
that hypoxia activates the ROCK2 pathway by upregulating ROCK2
expression and enhancing its activity in PAECs.
ROCK2 activity is blocked by Y27632
and ROCK2 siRNA significantly represses the expression of
ROCK2
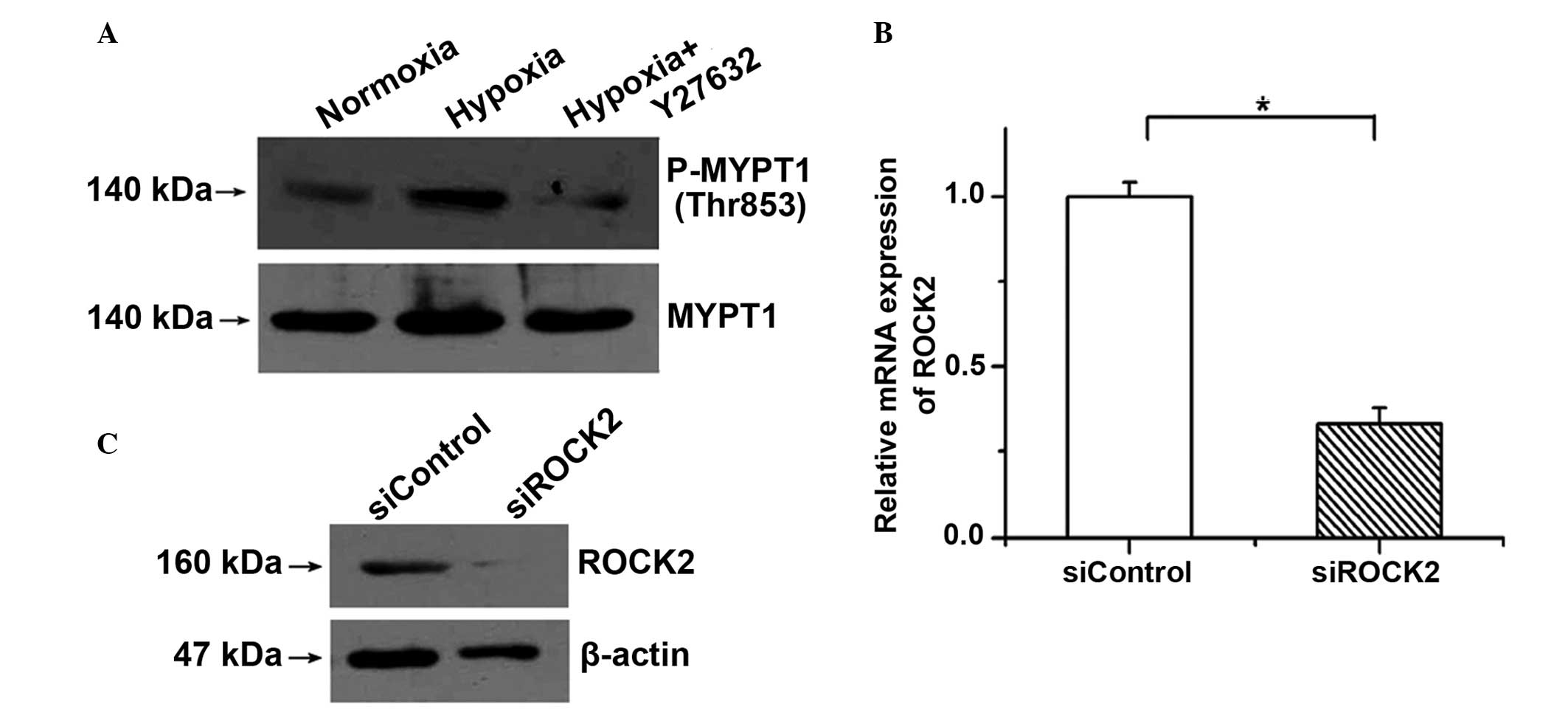
To demonstrate the important roles of ROCK2 in PAECs
treated with hypoxia, Y27632 (an inhibitor of ROCK) was used to
block the activation of the ROCK pathway. It was found that the
phosphorylation of MYPT1 induced by hypoxia was clearly decreased
by incubation with 1 µM Y27632 (Fig.
2A). As possible nonspecific inhibition by the chemical
inhibitor may occur, specific siRNA was also used to silence the
gene expression of ROCK2 in PAECs. The expression of ROCK2 was
examined by RT-qPCR and western blotting to determine the knockdown
efficiency. As shown in Fig. 2B and
C, the mRNA and protein expression of ROCK2 was significantly
reduced by siROCK2.
Hypoxia promotes the proliferation of
PAECs through the ROCK2 pathway
The MTT assay was conducted to determine whether the
effects of hypoxia on PAEC proliferation are dependent on the ROCK2
pathway. It was found that cell viability was increased by hypoxia
treatment, but the promotive effects of hypoxia on cell growth were
weakened by Y27632 (Fig. 3A; n=3,
P<0.05). Similar results were obtained after knocking down the
expression of ROCK2 with siROCK2; the cell viability increased by
hypoxia was repressed (Fig. 3B; n=3,
P<0.05). Furthermore, the results of the BrdU incorporation
assay showed that hypoxia increased the incorporation of BrdU into
PAECs, which was repressed by Y27632 or the ROCK2 siRNA (Fig. 3C and D; n=3, P<0.05). These
results indicate that the proliferation of PAECs induced by hypoxia
is mediated by ROCK2.
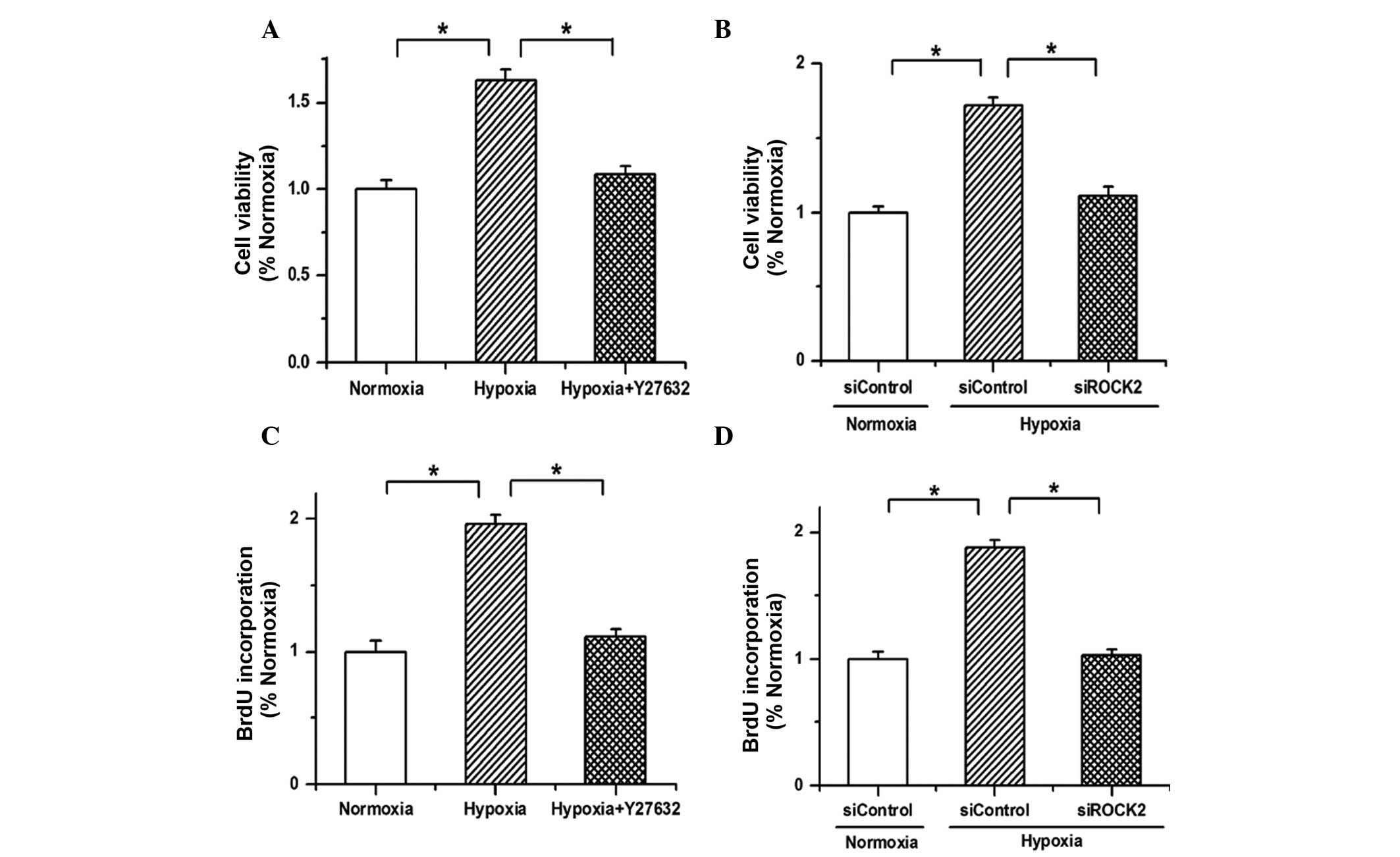 | Figure 3.Hypoxia promotes the proliferation of
PAECs through the ROCK2 pathway. (A) The results of an MTT assay
showed that the cell viability was enhanced by hypoxia treatment,
but the promotion effects of hypoxia on cell growth were weakened
by Y27632. (B) The increased cell viability of PAECs induced by
hypoxia was attenuated by ROCK2 siRNA (siROCK2). (C) Hypoxia
enhanced the incorporation of BrdU in PAECs, which was repressed by
Y27632. (D) The proliferation of PAECs promoted by hypoxia was
attenuated by siROCK2. All values are presented as the mean ±
standard error of the mean from three or more independent batches
of cells. *P<0.05, as detected by one-way analysis of variance
and Dunnett's test. PAECs, pulmonary arterial endothelial cells;
ROCK2, Rho-kinase α; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide;
siRNA, small interfering RNA; siControl, control siRNA; BrdU,
5-bromo-2′-deoxyuridine. |
Hypoxia regulates the expression of
cell cycle proteins to advance the cell cycle and its effects are
attenuated by siROCK2 or Y27632
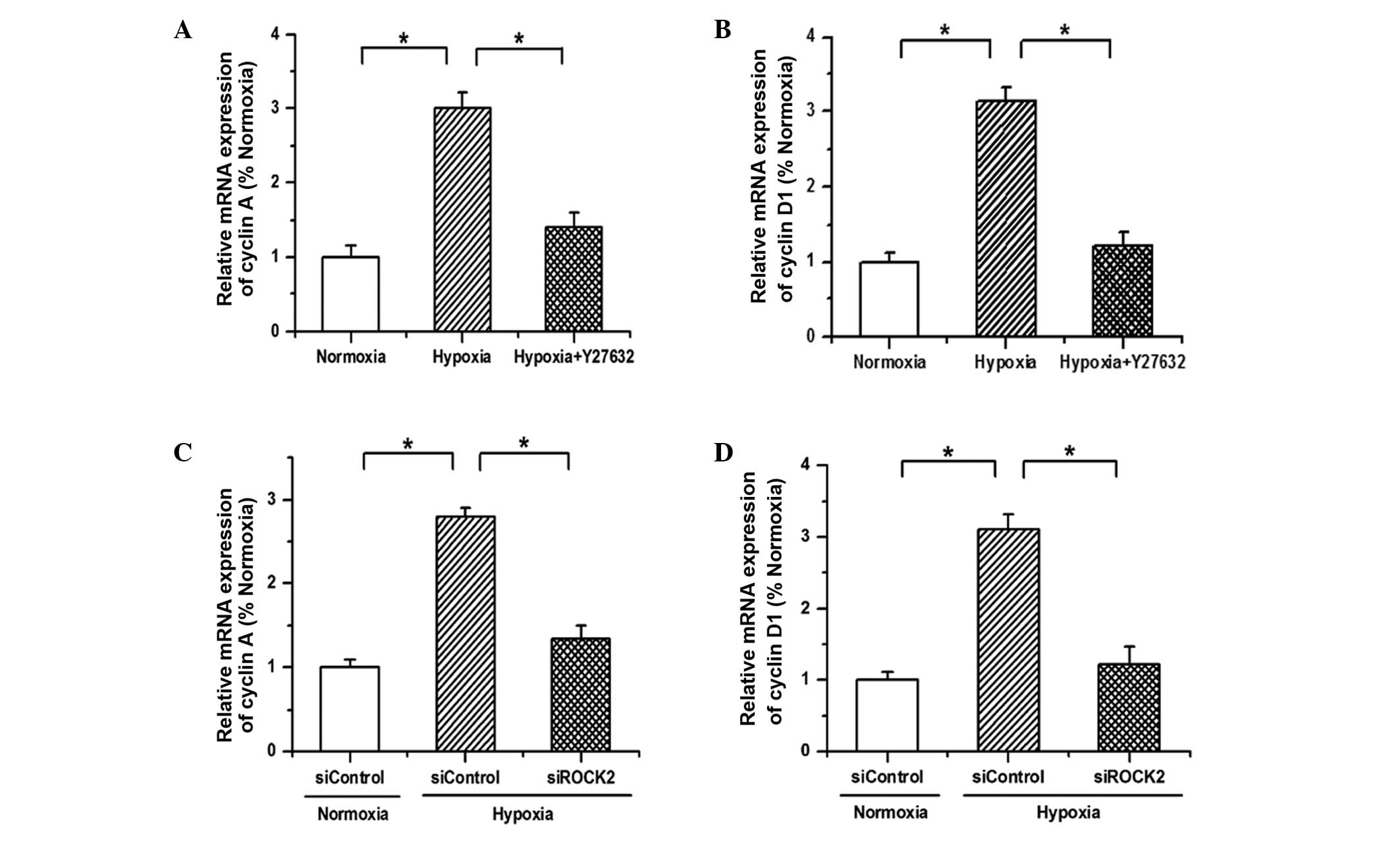
Since the aforementioned results have demonstrated
that hypoxia promotes the proliferation of PAECs via ROCK2, the
expression of cell-cycle regulatory proteins was then detected to
verify this conclusion. As shown in Fig.
4, the mRNA expression of cyclin A and cyclin D1 was
upregulated by hypoxia, and the promotive effects of hypoxia on the
expression of cyclin A and cyclin D1 were reversed after either
blocking the ROCK2 pathway with Y27632 or silencing the ROCK2
expression with siRNA. These results indicate that hypoxia promotes
the proliferation of PAECs by regulating the expression of cell
cycle proteins to advance cell cycle progression.
Discussion
Endothelial dysfunction is acknowledged as a common
pathological feature of PAH, which results from the formation of
plexiform lesions by the disordered overgrowth of endothelial cells
(14). Usually, inhibiting apoptosis
or promoting proliferation leads to the overgrowth of PAECs. The
overgrowth of pulmonary vascular endothelial cells is an important
feature of PAH, and the proliferated endothelial cells form
tumorlets and plexiform lesions, which can obliterate medium-sized
arteries and raise the pulmonary vascular pressure in the
development of PAH (14,15). Therefore, it is necessary to
determine the regulatory mechanism of proliferation in PAECs. The
results of the present study provide new evidence that ROCK2
mediates the proliferation of PAECs induced by hypoxia.
A major finding of the present study is that ROCK2
participates in the hypoxia-induced hypertrophy of the pulmonary
artery intima. There are two basic pathological changes in the
development of PAH: One is sustained pulmonary vasoconstriction and
the other is vascular remodeling. Previous studies have
demonstrated that the development of PAH induced by monocrotaline
or hypoxia is attenuated following a long-term treatment with
fasudil (an isoform-nonspecific ROCK inhibitor) (16,17).
Furthermore, increased ROCK activation has been observed in
patients with idiopathic PAH (18).
Moreover, it has been reported that the Rho/ROCK pathway is
involved in the regulation of pulmonary vasoconstriction (18,19).
This evidence indicates that the ROCK pathway serves as an
important signal transduction pathway in the progression of PAH.
However, the majority of studies have focused on the effects of
ROCK on the pulmonary vascular media (particularly pulmonary artery
smooth muscle cells) in blood vessel remodeling. Whether the ROCK
pathway is involved in the mechanism by which hypoxia induces
intimal hypertrophy remains unknown. As the major component of
vessel intima, PAECs were investigated used in the present study.
It was found that the activity and expression of ROCK2 were both
increased by hypoxia in cultured PAECs. Moreover, hypoxia promoted
the growth and proliferation of PAECs through activation of the
ROCK2 signaling pathway. These results provide direct evidence that
the effects of hypoxia on the overgrowth of PAECs are mediated by
ROCK2.
In addition, cyclin D1 and cyclin A are closely
associated with progression of the cell cycle. The increased
expression of cyclin D1 and cyclin A causes an increased number of
cells to transition from the G0/G1 phase to the S phase, and cell
proliferation is significantly enhanced (20). In the present study, hypoxia induced
the expression of cyclin D1 and cyclin A, but the ROCK inhibitor
Y27632 and ROCK2 siRNA each attenuated the effects of hypoxia on
the expression of cell cycle proteins. These results indicate that
hypoxia regulates the expression of cell cycle proteins to advance
cell cycle progression and promotes cell proliferation via
ROCK2.
In conclusion, the results of the present study
indicate that hypoxia advances the progression of the cell cycle
and promotes cell proliferation through activating the ROCK2
pathway in PAECs. This finding may highlight an important mechanism
in the development of PAH, and thus may suggest a novel target for
improving the treatment of PAH in the future.
References
|
1
|
Humbert M, Sitbon O, Chaouat A, Bertocchi
M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier JF, Chabot
F, et al: Survival in patients with idiopathic, familial and
anorexigen-associated pulmonary arterial hypertension in the modern
management era. Circulation. 122:156–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fukumoto Y and Shimokawa H: Recent
progress in the management of pulmonary hypertension. Circ J.
75:1801–1810. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morrell NW, Adnot S, Archer SL, Dupuis J,
Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA,
Weissmann N, et al: Cellular and molecular basis of pulmonary
arterial hypertension. J Am Coll Cardiol. 54(Suppl 1): S20–S31.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shimokawa H and Rashid M: Development of
Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol
Sci. 28:296–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Satoh K, Fukumoto Y and Shimokawa H:
Rho-kinase: Important new therapeutic target in cardiovascular
diseases. Am J Physiol Heart Circ Physiol. 301:H287–H296. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakagawa O, Fujisawa K, Ishizaki T, Saito
Y, Nakao K and Narumiya S: ROCK-I and ROCK-II, two isoforms of
rho-associated coiled-coil forming protein serine/threonine kinase
in mice. FEBS Lett. 392:189–193. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chevrier V, Piel M, Collomb N, Saoudi Y,
Frank R, Paintrand M, Narumiya S, Bornens M and Job D: The
rho-associated protein kinase p160ROCK is required for centrosome
positioning. J Cell Biol. 157:807–817. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei L, Roberts W, Wang L, Yamada M, Zhang
S, Zhao Z, Rivkees SA, Schwartz RJ and Imanaka-Yoshida K: Rho
kinases play an obligatory role in vertebrate embryonic
organogenesis. Development. 128:2953–2962. 2001.PubMed/NCBI
|
|
9
|
Shimizu Y, Thumkeo D, Keel J, Ishizaki T,
Oshima H, Oshima M, Noda Y, Matsumura F, Taketo MM and Narumiya S:
ROCK-I regulates closure of the eyelids and ventral body wall by
inducing assembly of actomyosin bundles. J Cell Biol. 168:941–953.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thumkeo D, Keel J, Ishizaki T, Hirose M,
Nonomura K, Oshima H, Oshima M, Taketo MM and Narumiya S: Targeted
disruption of the mouse rho-associated kinase 2 gene results in
intrauterine growth retardation and fetal death. Mol Cell Biol.
23:5043–5055. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Medhora M, Chen Y, Gruenloh S, Harland D,
Bodiga S, Zielonka J, Gebremedhin D, Gao Y, Falck JR, Anjaiah S and
Jacobs ER: 20-HETE increases superoxide production and activates
NAPDH oxidase in pulmonary artery endothelial cells. Am J Physiol
Lung Cell Mol Physiol. 294:L902–L911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma J, Zhang L, Li S, Liu S, Ma C, Li W,
Falck JR, Manthati VL, Reddy DS, Medhora M, et al:
8,9-Epoxyeicosatrienoic acid analog protects pulmonary artery
smooth muscle cells from apoptosis via ROCK pathway. Exp Cell Res.
316:2340–2353. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Budhiraja R, Tuder RM and Hassoun PM:
Endothelial dysfunction in pulmonary hypertension. Circulation.
109:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pidgeon GP, Tamosiuniene R, Chen G,
Leonard I, Belton O, Bradford A and Fitzgerald DJ: Intravascular
thrombosis after hypoxia-induced pulmonary hypertension: Regulation
by cyclooxygenase-2. Circulation. 110:2701–2707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abe K, Tawara S, Oi K, Hizume T, Uwatoku
T, Fukumoto Y, Kaibuchi K and Shimokawa H: Long-term inhibition of
Rho-kinase ameliorates hypoxia-induced pulmonary hypertension in
mice. J Cardiovasc Pharmacol. 48:280–285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abe K, Shimokawa H, Morikawa K, Uwatoku T,
Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K
and Takeshit A: Long-term treatment with a Rho-kinase inhibitor
improves monocrotaline-induced fatal pulmonary hypertension in
rats. Circ Res. 94:385–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doe Z, Fukumoto Y, Takaki A, Tawara S,
Ohashi J, Nakano M, Tada T, Saji K, Sugimura K, Fujita H, et al:
Evidence for Rho-kinase activation in patients with pulmonary
arterial hypertension. Circ J. 73:1731–1739. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oka M, Fagan KA, Jones PL and McMurtry IF:
Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary
hypertension. Br J Pharmacol. 155:444–454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Katula KS, Wright KL, Paul H, Surman DR,
Nuckolls FJ, Smith JW, Ting JP, Yates J and Cogswell JP:
Cyclin-dependent kinase activation and S-phase induction of the
cyclin B1 gene are linked through the CCAAT elements. Cell Growth
Differ. 8:811–820. 1997.PubMed/NCBI
|