Introduction
The World Health Organization calculates that ~171
million individuals worldwide have diabetes, and that its
prevalence will reach epidemic proportions by 2030, affecting ~366
million individuals (1). The
increasing prevalence of diabetes represents a significant burden
to human health due to its numerous and serious complications. It
has been suggested that chronic low-intensity inflammation and
insulin resistance (IR) are risk factors for type 2 diabetes
mellitus (T2DM), and are involved in the initial onset of T2DM and
the progression of its complications. Evidence from various
directions, including observational and experimental studies,
indicates the protective capacity of 1,25-dihydroxyvitamin D3
(active vitamin D) in chronic low-intensity inflammation and IR in
T2DM and its complications. Although the underlying mechanism is
yet to be clarified, it is recognized that this role may be at
least partially associated with the immunological regulation of
vitamin D, which is able to downregulate the production of
proinflammatory cytokines (2).
Previous studies have reported associations between vitamin D
insufficiency and diseases related to an amplified inflammatory
state, such as diabetes. The effects of vitamin D in downregulating
the proinflammatory profile in T2DM patients may be implicated in
the association between vitamin D deficiency and impaired glucose
tolerance or T2DM that has been observed in humans (3), and in the protective effects of vitamin
D supplementation (4).
Diabetes-induced liver complications are
characterized by varying degrees of inflammation, fatty
degeneration, fibrosis and other pathological changes. Current
studies regarding the effects of vitamin D on diabetes-induced
liver complications and its potential mechanisms in T2DM are seldom
reported. In consideration of the protective role of vitamin D
against inflammation and IR in T2DM, we hypothesized that the
cytokines or molecules involved in inflammation and IR in T2DM may
be targets of vitamin D. The present study was conducted to test
the hypothesis that vitamin D may improve the diabetes-induced
liver complications by suppressing the expression of inflammation
cytokines including C-Jun N-terminal kinase (JNK), C-Jun, tumor
necrosis factor (TNF)-α and interleukin (IL)-1β in rat models of
T2DM, thus determining the association, if any, between vitamin D,
inflammation and diabetes-induced liver complications.
Investigations into the targets of vitamin D may provide insight
into the potential therapeutic mechanism of diabetes-induced liver
complications.
Materials and methods
Animal modeling and grouping
A total of 34 male Sprague-Dawley rats (specific
pathogen-free; weight, 280±10 g; age, 8 weeks) were provided by the
Xinjiang Center for Disease Control and Prevention (Urumqi, China).
The rats were housed in individual cages maintained under ambient
temperature (25±2°C) and had ad libitum access to water and
food. After a week of acclimation, rats were randomly divided into
two groups: Normal control (NC; n=10) and T2DM model (n=24) groups.
The T2DM rat model was established by administering a high-fat and
high-sugar diet (containing 10% refined lard, 20% sucrose, 2%
cholesterol, 8% custard powder and 60% of normal diet; supplied by
the Institute of Research in Xinjiang Medical University, Urumqi,
China) for 8 weeks. Subsequently, the rats received a peritoneal
injection of 35 mg/kg streptozotocin (STZ; Sigma-Aldrich, St.
Louis, MO, USA) in 0.1 mol/l citrate buffer (pH 4.2; Weber Liyang
Chemical Group, Xi'an, China), while the NC group rats were fed the
basic diet and received citrate buffer alone. One week later,
random non-fasting blood glucose was measured from tail blood
samples using a portable glucometer (Accu-Chek, Mannheim, Germany).
Diabetes was determined by the presence of hyperglycemia (random
non-fasting glucose level, >16.7 mmol/l). Among the initial 24
rats in the T2DM model group, 22 met the hyperglycemia criteria.
Then, half of the T2DM model rats (n=12) were randomly allocated to
the vitamin D-treated group (VD; n=11), and each rat was
administered vitamin D (0.03 µg/kg/day; Shanghai Roche
Pharmaceutical Ltd., Shanghai, China) by filling the stomach using
a lavage needle (insertion depth, ~5 cm) for 8 weeks, while the
other 11 rats (T2DM group; DM) and the NC group received an
equivalent administration of peanut oil (Shandong Luhua Group Co.,
Ltd., Shandong, China) daily for 8 weeks. During the experimental
period, the NC group was fed the basic diet, while the DM and VD
groups were fed the high-fat and high-sugar diet. At the end of the
experiment, the NC and VD groups had retained 10 rats each, while 9
rats remained in the DM group. The study was approved by the ethics
committee of Xinjiang Medical University.
Tissue sampling and preparation
Following the trial, the rats were sacrificed using
2% sodium pentobarbital injection (50 mg/kg; Merck Millipore,
Darmstadt, Germany). The serum and liver specimens were harvested.
Blood samples were collected from the abdominal aorta and were
centrifuged at 990 × g for 20 min (5430R centrifuge; Eppendorf,
Hamburg, Germany) to separate the plasma for use in assays. The
serum levels of fasting plasma glucose (FPG) and fasting insulin
(FINS) were detected using a modular chemical analyzer (7600;
Hitachi, Tokyo, Japan) and an Architect i2000SR immunoassay
analyzer (Abbott Laboratories, Lake Bluff, IL, USA), respectively.
HOMA-IR was calculated using the following formula: (FPG ×
FINS)/22.5. Liver tissues were cut into small pieces, immersed into
RNAlater Stabilization Solution (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and stored at −80°C for subsequent reverse
transcription quantitative-polymerase chain reaction (RT-qPCR) and
western blot analyses. Remaining liver tissues were fixed in 4%
paraformaldehyde (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) and stored at 4°C for hematoxylin and eosin
(H&E; Shun Tian Biological, Ltd., Shanghai, China) staining and
immunohistology.
Histopathological staining
Fresh liver tissues were washed with saline, and
fixed in 4% paraformaldehyde. Following dehydration these tissues
were embedded in paraffin, and then cut into 5-µm sections using a
microtome (Leica Biosystems Nussloch GmbH, Nussloch, Germany).
Liver tissues from all rats were subjected to routine histological
examination. The sections were mounted on glass slides and depleted
of paraffin using xylene (Weber Liyang Chemical Group). Then,
sections were subjected to H&E staining. The sections were
observed using a digital image-capture system (DM300B; Leica
Microsystems).
Immunohistochemistry
Serial sections of 5 µm thickness were cut from
paraffin-embedded tissue blocks and placed onto glass slides. After
drying for 2 h, paraffin sections were deparaffinized and hydrated
through a series of graded alcohol. Endogenous peroxidase activity
was inactivated with 0.3% hydrogen peroxide. For antigen retrieval,
the glass slides were immersed in citrate buffer (0.01 M) at 95°C
for 20 min, and refrigerated at room temperature. Tissue slices
were incubated with rabbit anti-rat antibodies against C-Jun
(bs-0670R; 1:100; Beijing Biosynthesis Biotechnology, Co., Ltd.,
Beijing, China), JNK, TNF-α and IL-1β (BA1648, BA0131 and BA2782;
1:100; Wuhan Boster Biological Technology, Ltd., Wuhan, China)
respectively at 4°C overnight. Then secondary antibody (5:100;
ZLI-9017; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China) was added to incubate the slices for 20 min at room
temperature. After staining with 3,3′-diaminobenzidine chromogenic
reagent (Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.)
and counterstained with hematoxylin for 5–10 min. The slices were
sealed and visualized by microscopy using a digital image-capture
system (DM300B; Leica Microsystems). Brown staining was considered
to indicate positive detection. A negative control was performed by
omitting the primary antibody. The intensity of staining was
evaluated semi-quantitatively in five random microscopic fields
under high magnification (×400). The scoring criteria were based on
the percentage of positive area and the staining intensity. The
percentage of positive area was scored as follows: No positive
staining was scored as 0 points; <25% as 1 points; 25–50% as 2
points; 51–75% as 3 points; and >75% as 4 points. The staining
intensity was scored as 0–3 points, representing negative, weak,
moderate and strong staining, respectively. Five fields of each
section were evaluated and the average scores for each parameter
were calculated, then added. Zero points can be defined as negative
(−), 2–3 points were divided into weakly positive (+); 4–5 points
were divided into medium positive (++); and 6–7 points were divided
into strong positive (+++).
RT-qPCR
Total RNA was extracted using an RNeasy Mini kit
(Qiagen, Hilden, Germany). The concentrations of total RNA were
measured by absorbance at 260/280 nm (MD1000; Beijing Thmorgan
Biotechnology Co., Ltd., Beijing, China). Prior to reverse
transcription, gDNA eraser (Qiagen) was used to remove genomic DNA.
A QuantiTect Reverse Transcription Kit (Qiagen) was used to perform
reverse transcription using 14 µl RNA and oligo(dT) primers,
according to the manufacturer's instructions. The qPCR assays were
performed using an iQ5 Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using QuantiFast SYBR green
PCR Master Mix (Qiagen, Hilden, Germany) and 2 µl cDNA. The
sequences of the primers (designed by Sangon Biological Engineering
Co., Ltd., Shanghai, China) used were listed in Table I. A 20-µl reaction system was used
containing 10 µl master mix (Qiagen), 2 µl cDNA, 7.6 µl
H2O and 0.2 µl each of upstream and downstream primers.
The cycling conditions were as follows: Melt at 95°C for 10 min;
and 95°C for 10 sec, 60°C for 30 sec, for 40 cycles. Testing the
SYBR green I fluorescent intensity in the reaction process was used
to detect the increment of PCR products. A melting curve analysis
was used to confirm the specificity of the PCR product, which was
demonstrated as a single peak. The expression of β-actin served as
the internal reference. Every sample was analyzed in triplicate.
The relative expression levels of JNK, C-Jun, TNF-α and IL-1β were
calculated using the 2−ΔΔCq method (5).
 | Table I.Primers sequences for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primers sequences for reverse
transcription-quantitative polymerase chain reaction analysis.
| Primer | Sequence no. | Sequence (5′-3′) |
|---|
| JNK | NM-053829 | Forward:
GATTTGGTGTAGCCCTTGGA |
|
|
| Reverse:
GCTCACCCTTACCTGGAACA |
| C-Jun | NM-021835 | Forward:
GCACATCACCACTACTCCGA |
|
|
| Reverse:
GACACTGGGCAGCGTATT |
| TNF-α | NM-012675 | Forward:
TACTGAACTTCGGGGTGATCG |
|
|
| Reverse:
CCTTGTCCCTTGAAGAGAACC |
| IL-1β | NM-031512 | Forward:
TCCAGTCAGGCTTCCTTGTG |
|
|
| Reverse:
CGAGATGCTGCTGTGAGATT |
| β-actin | NM-031144 | Forward:
AGTACCCCATTGAACACGGC |
|
|
| Reverse:
TTTTCACGGTTGGCCTTAGG |
Western blot analysis
Total protein was extracted from liver tissues using
radioimmunoprecipitation assay (RIPA) buffer (containing 0.1%
PMSF), and the proteins were collected by centrifuging at 20,660 ×
g at 4°C for 10 min. As the protein phosphorylation was reversible
and would be dephosphorylated by phosphatase, there was need to
suppress or avoid cell phosphatase of endogenous and exogenous
disturbances in the process of sample preparation and the immune
detection, by using phosphatase inhibitor and protease inhibitor to
draw reliable conclusions. Phosphatase inhibitor and protease
inhibitor (Thermo Fisher Scientific, Inc.) were added to RIPA
buffer (Thermo Fisher Scientific, Inc.) according to manufacturer's
instructions. Protein concentrations were quantified using a
bicinchoninic protein assay kit with bovine serum albumin as the
standard (both purchased from Thermo Fisher Scientific, Inc.).
Equal quantities of protein (50 µg) were separated by 10% SDS-PAGE
(Beijing Solarbio Science & Technology, Co., Ltd.) and
electrotransferred to polyvinylidene fluoride membranes (Roche AG,
Basel, Switzerland). The membranes were blocked with 5% nonfat skim
milk in Tris-buffered saline containing 0.1% Tween 20 (TBST;
Beijing Solarbio Science & Technology Co., Ltd.) for 1 h at
room temperature and then incubated overnight at 4°C with
polyclonal rat anti-rabbit antibodies against JNK (cat. no. 9258S),
phospho-JNK (cat. no. 4671S), C-Jun (cat. no. 9165S) and
phospho-C-Jun (cat. no. 2361S) (all 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) then washed with TBST.
Following this, membranes were incubated with secondary anti-rabbit
antibody solution (1:1,000; cat. no. 1450236; Invitrogen; Thermo
Fisher Scientific, Inc.) for 2 h at room temperature, and washed
with TBST. Expression levels of the targeted proteins were detected
using BCIP/NBT chromogenic substrate (Invitrogen; Thermo Fisher
Scientific, Inc.). Protein concentrations were quantified using a
bicinchoninic protein assay kit. The intensity of each band was
captured digitally and measured using Quantity One software
(Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS software version 17.0 was used for statistical analysis (SPSS,
Inc., Chicago, IL, USA). Significant differences were determined
using one-way analysis of variance, and Least Significant
Difference testing was performed for pair-wise comparison. The
Wilcoxon Rank-Sum test was used for ranked data. P<0.01 was
considered to indicate a statistically significant difference.
Results
Effects of vitamin D on biochemical
parameters
The results of FPG, FINS and HOMA-IR are summarized
in Table II. Compared with the NC
group, the DM group exhibited increased levels of FPG, HOMA-IR and
decreased levels of FINS (P<0.01). In the VD group, FPG and
HOMA-IR were decreased, while FINS levels were increased compared
with the DM group (P<0.01).
| Table II.Biochemical parameters. |
Table II.
Biochemical parameters.
| Group | FPG (mmol/l) | FINS (mIU/l) | HOMA-IR |
|---|
| NC (n=10) |
5.80±0.94 | 19.49±1.53 |
5.01±0.81 |
| DM (n=9) |
24.66±2.47a |
11.81±1.36a |
12.86±1.33a |
| VD (n=10) |
20.39±2.12a,b |
13.07±1.15a,b |
11.77±0.80a,b |
| F-value | 153.214 | 59.724 | 78.73 |
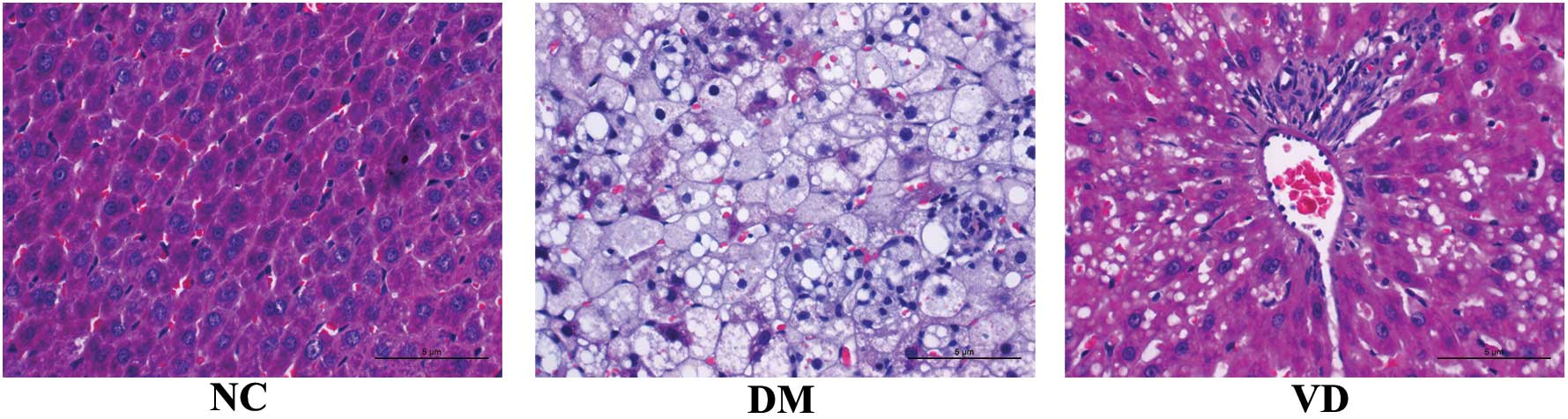
Effects of vitamin D on liver
histopathology
In the NC group, the structure of liver lobule was
normal and clear, the hepatic cord was orderly with a large, round
nucleus in the central of the hepatocyte and the cytoplasm is
uniform. The size of liver appeared larger by naked eye in DM group
compared with the NC group and was greasy to the touch. Various
pathological changes appeared in the DM group, including narrow
liver sinusoid, distortion of liver architecture, hepatocyte
swelling, fatty degeneration, cytoplasm rarefaction, spotty
necrosis scattering in the hepatic lobule as well as abundant
inflammatory cell infiltration. However, the changes described
above were markedly abated in the VD group (Fig. 1).
Immunohistochemical analysis of the
effects of vitamin D on liver and inflammatory cytokines
To investigate the molecular mechanisms underlying
the therapeutic effects of vitamin D on liver tissues in T2DM model
rats, immunohistochemical staining was performed to detect the
expression of JNK, C-Jun, TNF-α and IL-1β in the liver tissues. The
results showed that JNK, TNF-α and IL-1β were predominantly
expressed in the cytoplasm, while C-Jun was primarily expressed in
nucleus (Fig. 2). For JNK, C-Jun,
TNF-α and IL-1β there was obvious brown staining and their protein
expression levels were increased in the DM group compared with the
NC group (P<0.01). Conversely, compared with the DM group,
vitamin D administration significantly decreased the positive areas
and staining intensity of each target protein, and hence their
expression levels were significantly decreased in the VD group
(P<0.01) (Tables III and
IV).
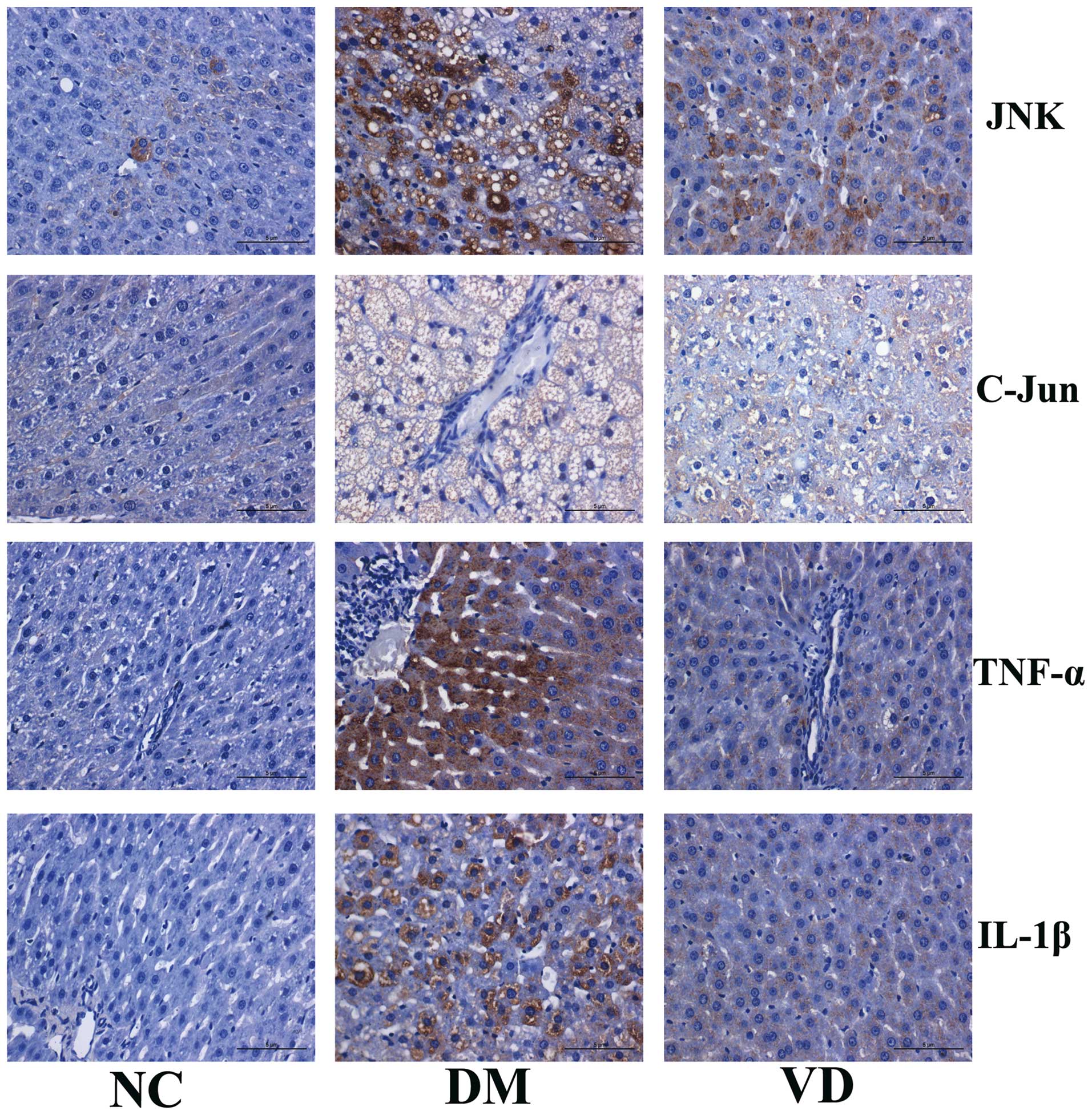 | Figure 2.Immunohistochemical analysis of the
protein expression of target genes in the liver tissues of type 2
diabetes mellitus model rats (magnification, ×400; stain,
hematoxylin and eosin). JNK, TNF-α and IL-1β are predominantly
expressed in the cytoplasm, while C-Jun is primarily expressed in
the nucleus. For JNK, C-Jun, TNF-α and IL-1β there is obvious brown
staining; protein expression levels are increased in the DM group
compared with the NC group. Conversely, compared with the DM group,
VD administration decreased the positive areas and staining
intensity of each target protein. JNK, c-Jun N-terminal kinase;
TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; NC, normal
control group; DM, type 2 diabetes mellitus group; VD, vitamin
D-treated group. |
| Table III.Immunohistochemical staining for JNK
and C-Jun protein expression levels in the rat liver tissues. |
Table III.
Immunohistochemical staining for JNK
and C-Jun protein expression levels in the rat liver tissues.
|
| JNK | C-Jun |
|---|
|
|
|
|
|---|
| Group | − | + | ++ | +++ | − | + | ++ | +++ |
|---|
| NC (n=10) | 8 | 2 | 0 | 0 | 6 | 4 | 0 | 0 |
| DM (n=9) | 0 | 1 | 2 | 6 | 0 | 0 | 1 | 8 |
| VD (n=10) | 1 | 4 | 3 | 2 | 0 | 4 | 3 | 3 |
| Table IV.Immunohistochemical staining for
TNF-α and IL-1β protein expression levels in the rat liver
tissues. |
Table IV.
Immunohistochemical staining for
TNF-α and IL-1β protein expression levels in the rat liver
tissues.
|
| TNF-α | IL-1β |
|---|
|
|
|
|
|---|
| Group | − | + | ++ | +++ | − | + | ++ | +++ |
|---|
| NC (n=10) | 7 | 3 | 0 | 0 | 8 | 2 | 0 | 0 |
| DM (n=9) | 0 | 0 | 3 | 6 | 0 | 1 | 1 | 7 |
| VD (n=10) | 1 | 4 | 4 | 1 | 2 | 3 | 4 | 1 |
RT-qPCR analysis of the effects of
vitamin D on the mRNA expression levels of inflammatory
cytokines
RT-qPCR was used to evaluate the changes in the mRNA
expression levels of the target genes in the liver tissues of the
T2DM model rats. The mRNA expression levels of JNK, C-Jun, TNF-α
and IL-1β in the NC group were relatively low, and were
significantly elevated in the DM group (P<0.01). Compared with
the DM group, vitamin D treatment appeared to significantly
decrease the expression levels of the target mRNAs (P<0.01)
(Table V).
| Table V.Reverse transcription-quantitative
polymerase chain reaction analysis of mRNA expression levels. |
Table V.
Reverse transcription-quantitative
polymerase chain reaction analysis of mRNA expression levels.
| Group | JNK | C-Jun | TNF-α | IL-1β |
|---|
| NC (n=10) | 1.02±0.11 | 0.90±0.12 | 0.82±0.09 | 0.77±0.09 |
| DM (n=9) |
7.39±1.27a |
5.15±0.98a |
6.14±1.13a |
6.31±1.33a |
| VD (n=10) |
5.93±1.29a,b |
3.76±0.97a,b |
4.78±1.07a,b |
5.09±0.84a,b |
| F-value | 99.707 | 72.906 | 62.305 | 55.018 |
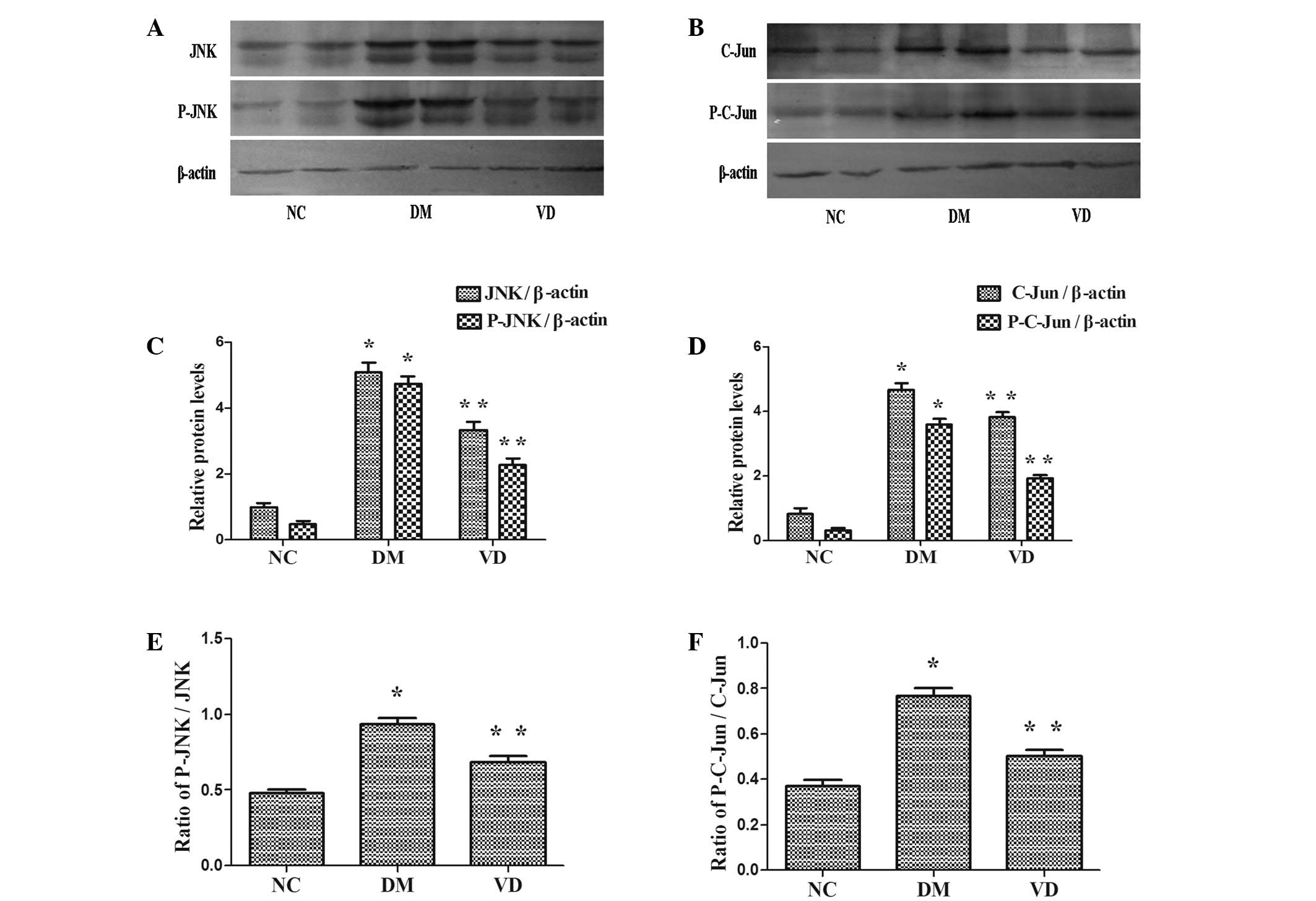
Western blot analysis of the effects
of vitamin D on the protein expression of inflammatory
cytokines
JNK is activated by phosphorylation and is able to
phosphorylate/activate the transcription factor C-Jun.
Consequently, the protein expression levels of JNK, phospho-JNK,
C-Jun and phospho-C-Jun were assessed using a western blot assay.
The protein expression levels of JNK, phospho-JNK, C-Jun and
phospho-C-Jun were higher in the DM group compared with the NC
group (P<0.01) (Fig. 3).
Furthermore, they were reduced in the VD group compared with the DM
group (P<0.01). Notably, the ratio of Phospho-JNK/JNK,
Phospho-C-Jun/C-Jun was identical to above descriptions in each
group (P<0.01).
Discussion
It has been demonstrated that inflammation and IR
play an initial role in the onset of T2DM, and that the progression
of its complications, suggesting that the regulation of
inflammation and IR may inhibit the emergence and progression of
this disease. Vitamin D is a well-known steroid hormone that has
been identified as a regulator of inflammation and IR. Based on
available clinical and epidemiological data, the positive effects
of vitamin D appear to be primarily related to its action on
inflammation and secondary to its action on insulin sensitivity and
secretion.
In the present study, H&E staining and
immunohistochemistry were performed to assess the direct effects of
vitamin D on diabetes-induced liver complications. The results
showed that the administration of vitamin D significantly
alleviated certain pathological changes, particularly steatosis and
inflammatory cell infiltration. Furthermore, immunohistochemistry
and RT-qPCR analysis showed that the expression levels of JNK,
C-Jun, TNF-α and IL-1β, which are implicated in inflammation and
IR, were decreased following treatment with vitamin D. Based on
these results, we speculate that vitamin D exerts therapeutic
effects on diabetes-induced liver complications, possibly by
downregulating the expression of JNK, C-Jun, TNF-α and IL-1β. This
modulation may be in part due to the anti-inflammatory properties
of vitamin D; however. the underlying mechanism remains to be
elucidated.
A previous study suggested that the activation of
inflammatory pathways interferes with normal metabolism and
disrupts insulin signaling (6). The
presence of chronic inflammatory diseases, such as rheumatoid
arthritis or hepatitis, significantly increases the risk for the
development of IR and/or T2DM, thus suggesting an association
between chronic inflammatory state and impaired insulin activity
(7,8). In addition, prior results have
indicated that vitamin D improves insulin sensitivity by its
anti-inflammatory activity. Incubation of isolated monocytes with
vitamin D attenuated the expression levels of proinflammatory
cytokines involved in IR, such as IL-1β, IL-6 and TNF-α in patients
with T2DM (9). Similarly in the
present study, in liver the tissues of T2DM model rats the levels
of IL-1β and TNF-α were decreased following the administration of
vitamin D, which was accompanied by an improvement in HOMA-IR.
Thus, we speculate that vitamin D is able to interfere with the
crosstalk between inflammation and IR.
Targher et al (10) demonstrated that the beneficial
effects of vitamin D were mediated through the modulation of VDR, a
receptor of vitamin D, thereby improving signal transduction in the
treatment of diabetes-induced liver complications. VDR is
constitutively expressed by immune cells, which suggests that
vitamin D plays an important role in the modulation of the
inflammatory response. The liver contains a large quantity of
Kupffer cells, which may be activated and release a wide range of
products implicated in liver injury, such as inflammatory cytokines
including TNF-α and IL-1β. These factors may further exacerbate
hepatic inflammation and IR, as found in the present study,
disabling the inflammatory pathway within these cells prevented the
generalization of inflammation and IR in liver (11). VDR is almost universally expressed in
nucleated cells (12), in which it
controls vital genes associated with bone metabolism, chronic
diseases and inflammation. Therefore, in the present study we
hypothesized that cells exhibiting VDR may be a target of vitamin
D. It has been demonstrated in monocytes, dendritic cells and T
cells that vitamin D is able to inhibit the expression of
inflammation-related genes by interacting with the NF-κB and
AP-1/C-Jun pathways (13). In the
present study, vitamin D appeared to downregulate the expression of
C-Jun and its upstream signaling molecule JNK in liver tissues.
JNKs were initially described as a family of
serine/threonine protein kinases, and may be activated by diverse
stimuli including cytokines (such as TNF-α and IL-1β), reactive
oxygen species, endoplasmic reticulum stress, free fatty acid,
hyperglycemia and advanced glycation end products, all of which are
known to be increased under diabetic conditions (14). JNK serves a crucial function in
chronic inflammatory diseases involving the expression of specific
proteases and cytokines (15). JNK
activity and/or expression levels are elevated in various tissues
in patients with T2DM, and have been shown to promote the
progression of inflammation and IR in T2DM and its complications,
which is in accordance with the present results. The specific
function of JNK in the dysregulation of hepatic insulin signaling
has also been demonstrated. Previous studies have indicated that
hepatic inhibition of JNK using a dominant-negative protein or
knockdown of JNK in diabetic mice significantly lowered blood
glucose levels and improved insulin sensitivity. Silencing of
hepatic JNK by small interfering RNA increased the expression of
glycolysis enzymes, reduced the expression of gluconeogenic enzymes
and the attenuated development of IR in a nutritionally-induced
diabetes mouse model (16). These
results directly demonstrate the therapeutic potential of JNK
inhibitors in diabetes. On the basis of the present results,
vitamin D may function as an inhibitor of JNK. Activated JNK is
able to directly target insulin receptor substrate 1 (IRS)-1 for
serine phosphorylation, which inhibits the insulin signaling
cascade (17). In addition, Sharfi
and Eldar-Finkelman (18) showed
that the expression level of JNK is markedly elevated in the
diabetic liver and that JNK/GSK-3-mediated phosphorylation of IRS-2
inhibited insulin signaling in liver cells. Thus, JNK has been
viewed as a potential target for the prevention and treatment of
T2DM (19).
At least 50 proteins have been identified as JNK
substrates. Among these substrates, C-Jun is a representative
target, as the functions of JNK depend largely on phosphorylating
C-Jun (20). Phosphorylated JNK
translocates into the nucleus and regulates downstream
transcriptional programs via transcription factor C-Jun, and
consequently promotes the activity of the activator protein-1
(AP-1). This promotion may result in the increased expression of
proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α. These
cytokines target cell membrane receptors, thus exacerbating the
inflammatory response and inducing IR (21). In addition to being leaner and more
insulin responsive, high-fat diet-fed JNK−/−
mice exhibited reduced expression of inflammatory cytokines such as
IL-6, TNF-α and MCP-1 (22).
Among the best-studied pathways leading to JNK
activation is TNF-α signaling via TNF-α receptor 1 (TNFR1)
(23). Mice that lack TNFR1 and rats
injected with an antibody against TNF-α exhibit reduced liver
activation of JNK and C-Jun/AP-1 (24). Notably, JNK expression has been
implicated in hepatocyte injury mediated by TNF-α (25). In hepatocytes, TNF-α rapidly
activates JNK, leading to the activation of C-Jun/AP-1 and
consequently the production of proinflammatory cytokines,
particularly TNF-α and IL-1β, which in return activate JNK as has
been noted. In conclusion, the interaction between JNK and TNF-α
forms a vicious circle and results in an increased quantity of
proinflammatory cytokines, which exacerbated inflammation and IR in
liver tissues in the present study. The administration of vitamin D
has been shown to inhibit the inflammatory response of diabetic
mice and reduce fasting glucose and serum TNF-α levels (26). Vitamin D administration also
inhibited the expression of TNF-α in monocytes extracted from
diabetic patients (9). Furthermore,
TNF-α is hypothesized to play a major role in the pathophysiology
of IR in rodents (27) via the
phosphorylation of IRS-1 protein on serine residues. Recombinant
TNF-α decreases insulin sensitivity, while TNF-α or TNF-α receptor
null mice exhibit an increased sensitivity in response to this
hormone, suggesting a direct role for TNF-α in the development of
IR.
In chronic liver disease, the overproduction of
IL-1β and TNF-α is observed (28).
Negrin et al (29) reported
that the development of hepatic steatosis required IL-1β signaling,
which upregulated fatty acid synthase to promote hepatic
lipogenesis. Furthermore, the administration of IL-1β receptor
antagonist to obese mice markedly reduced steatosis in hepatic
tissues. IL-1β binding to IL-1β receptor I (IL-1RI) activated a
number of inflammatory pathways, including JNK, which induced IR by
attenuating IRS-1 activation while activating C-Jun/AP-1 and
consequently inducing the transcription of inflammatory genes
including itself (24), similarly to
TNF-α, ultimately resulting in extensive inflammation reactions in
the liver. IL-1RI−/− were protected against
high-fat diet-induced IR. Recent clinical studies aimed at
attenuating IL-1β activity in subjects with T2DM have yielded
promising results; demonstrating improvements in hyperglycaemia,
insulin secretion and sensitivity, β-cell function and reduced
levels of systemic inflammation (30). Moore et al (31) suggested that the anti-inflammatory
effect of vitamin D3 derived from its ability to downregulate the
IL-1β-induced activation of JNK in microglial cells.
It is currently unclear whether JNK is the cause or
effect of other cytokines, particularly TNF-α and IL-1β. The
targeting of individual inflammatory cytokines may not be highly
effective, whereas the targeting of inflammatory kinases JNK
generated a robust anti-diabetic action as this factor integrated
signals from multiple inflammatory mediators. Therefore, targeting
JNK could potentially disable the inflammatory response and improve
insulin action (32). For example,
TNF-α is a key downstream effectors of JNK-mediated IR in 3T3-L1
adipocytes (33). In the present
study, the protein expression levels of JNK and C-Jun, in addition
to their active forms phospho-JNK and phospho-C-Jun, were evaluated
using western blot assays. The results showed that compared with
the DM group, vitamin D was able to downregulate the protein
expression of JNK and C-Jun, their active forms and the ratios of
phospho-JNK/JNK and phospho-C-Jun/C-Jun. These results suggested
that vitamin D was able to regulate the phosphorylation of JNK and
C-Jun. Notably, increased dietary vitamin D was beneficial in
preventing inflammation-associated colon cancer via the suppression
of inflammatory responses, particularly JNK (34). Supplementation with vitamin D
improved IR in muscle cells by reducing the phosphorylation of JNK
(35). These results suggest that
vitamin D has a crucial impact on inflammation and IR induced by
JNK. Thus, JNK signaling pathway may be a crucial target of vitamin
D with regarding to anti-inflammation and improves IR in
diabetes-induced liver complications. Hence, the inhibition of the
JNK pathway prevents its activation by various stimuli,
particularly TNF-α and IL-1β, and contributes to the reduced
production of proinflammatory cytokines in the liver. In
conclusion, vitamin D may play a role in attenuating the positive
feedback loop between JNK signaling pathway and TNF-α and IL-1β,
thus ameliorating the diabetes-induce liver complications in T2DM
model rats.
Lipid infusion or high fat feeding promotes IR in
rodents and humans (33). IR occurs
in the liver and other tissues, resulting in metabolism disturbance
which may result in the accumulation of fat in hepatocytes.
Notably, the Third National Health and Nutrition Examination Survey
identified an inverse association between vitamin D status and
IR/diabetes (36). Significant
improvements were observed in insulin sensitivity and IR in a
randomized, controlled, double-blind intervention, which involved
the administration of 100 µg vitamin D (4,000 IU) for 6 months to
South Asian women who were insulin resistant (HOMA-IR, >1.93;
serum 25(OH)D, <50 nmol/l) (37),
which was in agreement with the present findings. In the present
study, the HOMA-IR in the DM group was significant higher compared
with the NC group following vitamin D treatment, thus showing a
tendency toward improvement. Although the underlying mechanisms
remain unclear, it may partly due to the anti-inflammatory
properties of vitamin D. The JNK-C-Jun axes is a major inflammatory
pathway involved in the disruption of insulin signaling, and
modulating its action with anti-inflammatory agent is believed to
improve insulin sensitivity and glucose homeostasis (38). In the present study, vitamin D may
have served as the anti-inflammatory agent in regulating JNK/C-Jun,
TNF-α and IL-1β in insulin signaling.
It has been reported that the combination of
lipotoxicity and glucotoxicity induced by a high-fat diet and STZ
injection leads to islet β-cell failure and apoptosis, and
ultimately to hypoinsulinemia and hyperglycemia (39). This is similar to the present method
of T2DM model establishment, and closely mimics the natural history
and metabolic characteristics of late stage T2DM. T2DM is
characterized by an increase in IR in conjunction with the
inability of islet β-cells to secrete sufficient insulin to
compensate. IR and hypoinsulinemia may result in a hyperglycemic
state that is a major risk factor for the development of
diabetes-induced liver complications. Hyperglycemia promotes the
accumulation of fat in the liver, while releases oxidizing and
inflammatory mediators and causing local and systemic damage. In
the present study, the administration of vitamin D reduced the FPG
and increased the FINS. These changes are in accordance with a
prior study (40) which showed that
vitamin D is able to act on β-cells through the vitamin D response
elements present in insulin gene promoter, thereby increasing the
levels of FINS in plasma which may result in decreased FBG levels
in vitamin D-treated group. In addition, the immunomodulatory
properties of vitamin D may decrease the release of inflammatory
cytokines such as TNF-α and IL-1β, while improving IR-induced by
inflammation, thereby restoring residual β-cells and decreasing FPG
induced by IR.
In summary, the present results indicate the
protective effects of vitamin D against diabetes-induced liver
complications, which may occur by modulating the inflammatory
response, attenuating the crosstalk between inflammation and IR and
ameliorating hyperglycemic state. The results suggest an
association between vitamin D, inflammation and T2DM.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81160116).
References
|
1
|
Guadarrama-López AL, Valdés-Ramos R and
Martínez-Carrillo BE: Type 2 diabetes, PUFAs, and vitamin D: Their
relation to inflammation. J Immunol Res. 2014:8607032014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Flores M: A role of vitamin D in
low-intensity chronic inflammation and insulin resistance in type 2
diabetes mellitus? Nutr Res Rev. 18:175–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chiu KC, Chu A, Go VL and Saad MF:
Hypovitaminosis D is associated with insulin resistance and beta
cell dysfunction. Am J Clin Nutr. 79:820–825. 2004.PubMed/NCBI
|
|
4
|
Pittas AG, Dawson-Hughes B, Li T, Van Dam
RM, Willett WC, Manson JE and Hu FB: Vitamin D and calcium intake
in relation to type 2 diabetes in women. Diabetes Care. 29:650–656.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gregor MF and Hotamisligil GS:
Inflammatory mechanisms in obesity. Ann Rev Immunol. 29:415–445.
2011. View Article : Google Scholar
|
|
7
|
Sattar N, McCarey DW, Capell H and McInnes
IB: ‘Explaining how high-grade’ systemic inflammation accelerates
vascular risk in rheumatoid arthritis. Circulation. 108:2957–2963.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Knobler H, Zhornicky T, Sandler A, Haran
N, Ashur Y and Schattner A: Tumor necrosis factor-alpha-induced
insulin resistance may mediate the hepatitis C virus-diabetes
association. Am J Gastroenterol. 98:2751–2756. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giulietti A, van Etten E, Overbergh L,
Stoffels K, Bouillon R and Mathieu C: Monocytes from type 2
diabetic patients have a pro-inflammatory profile.
1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res
Clin Pract. 77:47–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Targher G, Bertolini L, Scala L, Cigolini
M, Zenari L, Falezza G and Arcaro G: Associations between serum
25-hydroxyvitamin D3 concentrations and liver histology in patients
with non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis.
17:517–524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laskin DL, Weinberger B and Laskin JD:
Functional heterogeneity in liver and lung macrophages. J Leukoc
Biol. 70:163–170. 2001.PubMed/NCBI
|
|
12
|
Korf H, Wenes M, Stijlemans B, Takiishi T,
Robert S, Miani M, Eizirik DL, Gysemans C and Mathieu C:
1,25-Dihydroxyvitamin D3 curtails the inflammatory and T cell
stimulatory capacity of macrophages through an IL-10-dependent
mechanism. Immunobiology. 217:1292–1300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alroy I, Towers TL and Freedman LP:
Transcriptional repression of the interleukin-2 gene by vitamin D3:
Direct inhibition of NFATp/AP-1 complex formation by a nuclear
hormone receptor. Mol Cell Biol. 15:5789–5799. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ip YT and Davis RJ: Signal transduction by
the c-Jun N-terminal kinase (JNK)-from inflammation to development.
Curr Opin Cell Biol. 10:205–219. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karin M and Gallagher E: From JNK to pay
dirt: Jun kinases, their biochemistry, physiology and clinical
importance. IUBMB Life. 57:283–295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakatani Y, Kaneto H, Kawamori D, Hatazaki
M, Miyatsuka T, Matsuoka TA, Kajimoto Y, Matsuhisa M, Yamasaki Y
and Hori M: Modulation of the JNK pathway in liver affects insulin
resistance status. J Biol Chem. 279:45803–45809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang R and Trevillyan JM: c-Jun N-terminal
kinase pathways in diabetes. Int J Biochem Cell Biol. 40:2702–2706.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sharfi H and Eldar-Finkelman H: Sequential
phosphorylation of insulin receptor substrate-2 by glycogen
synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in
hepatic insulin signaling. Am J Physiol Endocrinol Metab.
294:E307–E315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaneto H: The JNK pathway as a therapeutic
target for diabetes. Expert Opin Ther Targets. 9:581–592. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tarantino G and Caputi A: JNKs, insulin
resistance and inflammation: A possible link between NAFLD and
coronary artery disease. World J Gastroenterol. 17:3785–3794. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chagas CE, Borges MC, Martini LA and
Rogero MM: Focus on vitamin D, inflammation and type 2 diabetes.
Nutrients. 4:52–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tuncman G, Hirosumi J, Solinas G, Chang L,
Karin M and Hotamisligil GS: Functional in vivo interactions
between JNK1 and JNK2 isoforms in obesity and insulin resistance.
Proc Natl Acad Sci USA. 103:10741–10746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schwabe RF, Uchinami H, Qian T, Bennett
BL, Lemasters JJ and Brenner DA: Differential requirement for c-Jun
NH2-terminal kinase in TNF alpha-and Fas-mediated apoptosis in
hepatocytes. FASEB J. 18:720–722. 2004.PubMed/NCBI
|
|
26
|
Li H, Xie H, Fu M, Li W, Guo B, Ding Y and
Wang Q: 25-hydroxyvitamin D3 ameliorates periodontitis by
modulating the expression of inflammation-associated factors in
diabetic mice. Steroids. 78:115–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-alpha:
Direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bieghs V and Trautwein C: The innate
immune response during liver inflammation and metabolic disease.
Trends Immunol. 34:446–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Negrin KA, Roth Flach RJ, Di Stefano MT,
Matevossian A, Friedline RH, Jung D, Kim JK and Czech MP: IL-1
signaling in obesity-induced hepatic lipogenesis and steatosis.
PloS One. 9:e1072652014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Larsen CM, Faulenbach M, Vaag A, Vølund A,
Ehses JA, Seifert B, Mandrup-Poulsen T and Donath MY:
Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N
Engl J Med. 356:1517–1526. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moore M, Piazza A, Nolan Y and Lynch MA:
Treatment with dexamethasone and vitamin D3 attenuates
neuroinflammatory age-related changes in rat hippocampus. Synapse.
61:851–861. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nguyen MT, Satoh H, Favelyukis S,
Babendure JL, Imamura T, Sbodio JI, Zalevsky J, Dahiyat BI, Chi NW
and Olefsky JM: JNK and tumor necrosis factor-alpha mediate free
fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol
Chem. 280:35361–35371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meeker S, Seamons A, Paik J, Treuting PM,
Brabb T, Grady WM and Maggio-Price L: Increased dietary vitamin D
suppresses MAPK signaling, colitis, and colon cancer. Cancer Res.
74:4398–4408. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou QG, Hou FF, Guo ZJ, Liang M, Wang GB
and Zhang X: 1,25-Dihydroxyvitamin D improved the free
fatty-acid-induced insulin resistance in cultured C2C12 cells.
Diabetes Metab Res Rev. 24:459–464. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Scragg R, Sowers M and Bell C: Third
National Health and Nutrition Examination Survey: Serum
25-hydroxyvitamin D, diabetes, and ethnicity in the third national
health and nutrition examination survey. Diabetes Care.
27:2813–2818. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
von Hurst PR, Stonehouse W and Coad J:
Vitamin D supplementation reduces insulin resistance in South Asian
women living in New Zealand who are insulin resistant and vitamin D
deficient-a randomised, placebo-controlled trial. Br J Nutr.
103:549–555. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nugent DA, Smith DM and Jones HB: A review
of islet of Langerhans degeneration in rodent models of type 2
diabetes. Toxicol Pathol. 36:529–551. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Meerza D, Naseem I and Ahmed J: Effect of
1,25(OH)2 vitamin D3 on glucose homeostasis
and DNA damage in type 2 diabetic mice. J Diabetes Complications.
26:363–368. 2012. View Article : Google Scholar : PubMed/NCBI
|