Introduction
3-Hydroxy-3-methylglutaric aciduria (3-HMG), also
called 3-hydroxy-3-methylglutaryl-CoA lyase (HMG-CoA lyase or HL)
deficiency, is an autosomal recessive disorder of the metabolism
(1). This disease was first
described in 1976 in a 7-month old boy from Australia (2). His major symptoms included vomiting,
cyanosis, apnea, metabolic acidosis, hypoglycemia, and an increase
in urine 3-hydroxy-3-methylglutaric acid (2). To date, ~100 cases have been reported
worldwide, with an incidence of <1/100,000 among live newborns
(3). The highest prevalence of the
disease is in Saudi Arabia (3), and
it is rarely observed in other populations, with only two reports
from Taiwan and two from mainland China (4,5).
HL catalyzes the final step of the mitochondrial
ketogenic pathway and leucine catabolism by cleaving HMG-CoA to
yield acetyl-CoA and acetoacetic acid (6). In the absence of HL, leucine cleavage
is incomplete, resulting in accumulation of metabolic acids and
compromised ketone body synthesis (6). Elevated levels of metabolic acids often
cause liver damage and/or hepatomegaly in 3-HMG patients (7). Normally, during periods of fasting or
infection, generation of ketone bodies in mitochondria provide an
alternative energy source for the brain, heart and kidney (6). Impairments in ketogenesis may lead to
hyperammonemia, and hypoketotic hypoglycemia can result in acute
brain damage, apnea and in certain cases sudden fatality (8).
Few reports have assessed brain damage in 3-HMG
patients using magnetic resonance imaging (MRI). Among these,
predominantly cerebral white matter alternations were described
(9,10). The present study reports the case of
a patient with a particularly severe case of 3-HMG with prominent
bilateral basal ganglia damage.
Case report
All procedures followed were in accordance with the
ethical standards of the responsible committee on human
experimentation of The First Hospital of Jilin University
(Changchun, China) and with the Helsinki Declaration of 1975, as
revised in 2000 (7). Informed
consent was obtained from the parents of the patient.
A 20-month old male was admitted to The First
Hospital of Jilin University with colic lasting for 3 days and
onset of intermittent seizures. Although the parents reported fever
of the patient, the temperature was not measured. Seizures
presented as loss of consciousness, cyanosis and muscle stiffness.
Five seizures were reported, and each seizure lasted 5–6 min. Prior
to admission, the patient's symptoms were alleviated and
consciousness was regained. The patient was the only pregnancy of
his mother, and was delivered at full term without complications.
At birth, the patient weighed 3.5 kg, with a head circumference of
36 cm and no birth asphyxia. The child was formula-fed, and
suffered from recurrent diarrhea and was frequently hospitalized,
averaging once per month. The boy had a poor appetite and had
iron-deficient anemia (hemoglobin, 95–105 g/l; reference, 120–160
g/l). Regarding physical growth and neurological development, the
patient was 2–3 months delayed behind children of similar age. The
patient was able to walk independently and call for his parents by
name. There was no family history of inherited disease or
hepatitis.
At the time of hospitalization, the 20-month old was
82 cm tall, weighing 10 kg and with a head circumference of 52 cm.
On physical examination, the patient exhibited thin hair and pale
skin. Patient was in a semi-coma, had slow pupillary reflex to
light and slightly congested throat. The patients muscle tension
and knee reflex were weak, and the Babinski sign was negative. No
abnormalities in the heart, lungs and abdomen were discerned.
During the first week of hospitalization, the
patient experienced intermittent fever, averaging two or three
episodes per day, with a maximum temperature of 39°C. Seizures
occurred one to two times per days with symptoms as described
previously. Blood samples were collected from the child with his
parents' informed consent. Routine blood tests reported: White
blood cell count, 8.24×109/l; neutrophil percentage,
0.84%; lymphocyte percentage, 0.11%; red blood cell count,
3.89×1012/l; hemoglobin, 86 g/l; hematocrit, 0.278 l/l;
mean corpuscular volume, 71.5 fl; high sensitive C-reactive
protein, 23 mg/l (reference, 0–3 mg/l); procalcitonin (PCT), 54.20
ng/ml (reference, 0–0.5 ng/ml); and urinary ketone (−), iron 7.3
mmol/l (reference, 9–22 mmol/l). Ferritin, total iron binding
capacity, folic acid and vitamin B12 were all within the
normal range. Blood gas analysis showed metabolic acidosis,
hyperammonemia, hypoglycemia and increased liver enzyme activity
(Table I). HMG-CoA lyase activity in
lymphocytes (Synergy H1; BioTeke Corporation, Beijing, China) was
~5% of the normal activity. Ceruloplasmin level was normal, as was
a routine cerebrospinal fluid biochemical test (UniCel®
DxC 800 Synchron® Clinical System; Beckman Coulter,
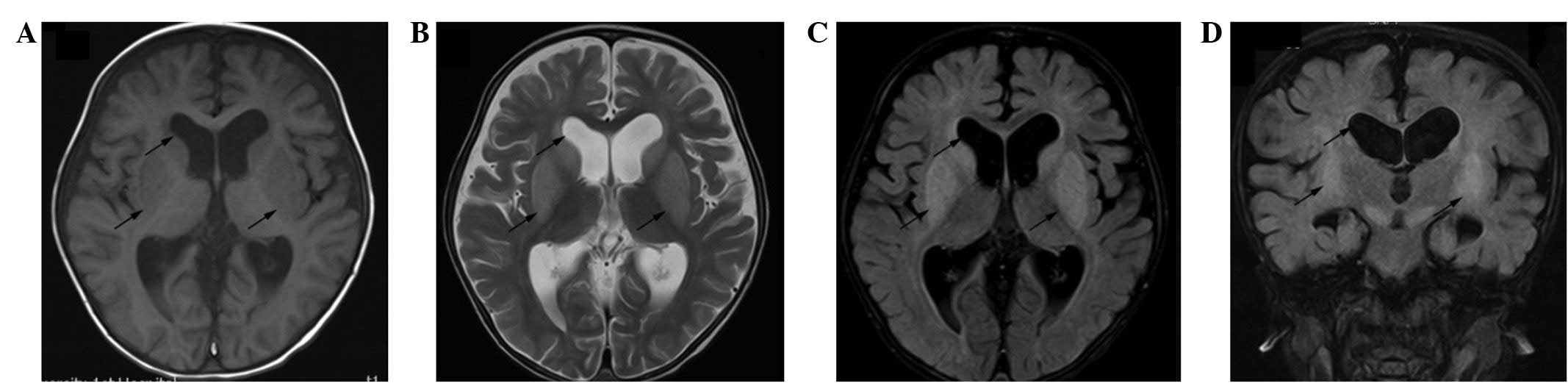
Inc., Brea, CA, USA). Results from brain axial MRI (MAGNETOM
Avanto; Siemens AG, Munich, Germany) are shown in Fig. 1. White matter abnormalities were
observed in the bilateral frontotemporal lobe and ventricle. In
T1-weighted, T2-weighted (Fig. 1A and
B), and fluid-attenuated inversion recovery images (Fig. 1C and D), marked symmetrical and
bilateral patchy and focal signals in the brain stem and basal
ganglia were detected (Fig. 1C and
D). The ventricles were enlarged, with a widening of the groove
of the anterior temporal, and parietal lobes, bilaterally (Fig. 1, upper arrow). The most prominent
abnormality was a bilateral hyperintensive signal in the basal
ganglia (Fig. 1B–D, lower arrows).
Metabolic profiling of urine was performed using gas
chromatography/mass spectrometry (GC/MS) analysis (GCMS-QP2010
Ultra; Shimadzu Corporation, Kyoto, Japan). Levels of
3-hydroxy-3-methyl-glutaric acid, 3-methyl-glutaric acid,
3-methyl-glutaconic acid, 3-hydroxy-isovaleric acid,
isovaleric-glycine, 3-methyl-crotonyl-glycine, glutaric acid and
hexane diacid were increased (Table
II). Tandem MS analysis of blood (API 3200MD; AB SCIEX,
Framingham, MA, USA) identified increased
3-hydroxy-isovaleryl-carnitine and adipoyl-carnitine levels
(Table II) and normal amino acid
levels. Based on the above findings, the patient was diagnosed with
3-HMG, sepsis, metabolic acidosis, and iron-deficient anemia, and
was prescribed cefepime dihydrochloride monohydrate (0.5 g twice
daily intravenous injection; Youcare Pharmaceutical Group Co.,
Ltd., Beijing, China), levetiracetam (0.125 g Keppra twice daily;
UCB Pharma S.A., Braine-l'Alleud, Belgium) and L-carnitine (0.5 ml
Levocarnitine Oral Solution twice daily; Northeast Pharmaceutical
Group Co., Ltd., Shenyang, China).
 | Table I.Biochemical analyses of the patient at
different time points of hospitalization. |
Table I.
Biochemical analyses of the patient at
different time points of hospitalization.
| Parameter | Hospital
admission | 2 weeks after
hospitalization | 6 months after
hospitalization | Reference range |
|---|
| Blood pH | 7.34 | 7.38 | 7.41 | 7.35–7.45 |
|
HCO3− (mmol/l) | 14 | 23 | 25 | 21–28 |
| Base excess
(mmol/l) | −11.8 | −2.5 | −2 | −3–3 |
| Lactic acid
(µmol/l) | 1.5 | 0.9 | 0.3 | 0.5–1.6 |
| Blood ammonia
(µmol/l) | 90 | 41 | 78 | 9–47 |
| Aspartate
transaminase (U/l) | 156 | 120 | 92 | 15–40 |
| Alanine
aminotransferase (U/l) | 145 | 107 | 191 | 9–50 |
| Glucose (mmol/l) | 3.7 | 4.8 | 4.5 | 3.9–6.1 |
| Table II.Urine gas chromatography/mass
spectrometry and blood tandem mass spectrometry analysis. |
Table II.
Urine gas chromatography/mass
spectrometry and blood tandem mass spectrometry analysis.
| Analyte | Value | Reference |
|---|
| Urinary acid (mg/g
creatinine) |
|
|
|
3-Hydroxy-3-methyl-glutaric
acid | 655.77 | 0–25.7 |
|
3-Methyl-glutaric acid | 269.67 | 0–4.5 |
|
3-Methyl-glutaconic acid | 615.28 | 0–4.2 |
|
3-Hydroxy-isovaleric acid | 413.45 | 0–2.3 |
|
Isovaleric-glycine | 1.73 | 0–0.4 |
|
3-Methyl-crotonyl-glycine |
117.34 | 0–0 |
| Glutaric
acid |
44.96 | 0–4 |
| Hexane
diacid |
71.82 | 0.5–5 |
| Blood carnitine
spectrum (µmol/l) |
|
|
|
3-Hydroxy-isovaleryl-carnitine | 5.04 | 0.06–0.6 |
|
Adipoyl-carnitine | 0.09 | 0–0.06 |
The patient's symptoms improved after 15 days of
hospitalization. However, the patient continued to exhibit weak
response to stimuli, unresponsiveness to calling and opisthotonos.
Patient was maintained on a liquid diet and had no fever, vomiting,
diarrhea or convulsions. Neurological examination revealed soft
neck, normal muscle strength in limbs, increased muscle tension,
normal knee reflex and positive Babinski sign. Blood tests showed
normal levels of high sensitive C-reactive protein and PCT. Blood
gas analysis showed that blood ammonia and glucose levels were
normal. Liver aminotransferase activity remained elevated (Table I). The patient was discharged, and
leucine-free formula and L-carnitine were prescribed. This
treatment regimen, however, was not administered by the parents
after discharge.
At the 6 month follow-up, the patient continued to
suffer from intermittent vomiting and diarrhea. He could open and
close his eyes, but was bedridden, unresponsive to calling, had
unconscious limb movement and high muscle tension. The results of a
blood gas analysis and the blood sugar level were normal, although
liver aminotransferase activity and blood ammonia levels remained
high (Table II). At the 1 year
follow-up, the patient showed no significant changes and the
parents did not agree to conducting review laboratory tests and MRI
for economic reasons.
Discussion
The onset of 3-HMG varies between individuals, with
~30% of patients displaying symptoms at the neonatal stage, while
the remaining 60–70% of patients develop symptoms between 3 and 12
months after birth (11). These
patients often present clinically with the following symptoms:
Vomiting, diarrhea, hypotonia, hypothermia, lethargy, apnea and
coma (12). The lethality rate for
3-HMG is ~20%. Since early manifestations of the disease lack
specificity and routine laboratory tests often fail to detect
abnormality when the disease is in remission, 3-HMG is frequently
misdiagnosed as septicemia or hypoglycemia (12). Diagnosis of 3-HMG requires GC/MS
profiling of organic acids in urine and tandem MS analysis of blood
(8). An increase in
3-hydroxy-isovaleryl-carnitine and adipoyl-carnitine in tandem MS
analysis of blood and an increase in urine secretion of
3-hydroxy-3-methyl-glutaric acid, 3-methyl-glutaric acid,
3-hydroxy-isovaleric acid, and 3-methyl-glutaconic acid are
indicative of 3-HMG (13). Among
these, 3-hydroxy-3-methyl-glutaric acid is a specific marker for
3-HMG. The gene encoding HL is on chromosome lp36.1-p35 and
consists of nine exons and eight introns. To date, >50 point
mutations identified over the entire HL gene have been described
(10). Therefore, DNA sequencing may
be used to confirm a diagnosis of 3-HMG. In addition, other
diagnostic methods include a skin fibrosis test and an HL activity
test in lymphocytes or from a liver biopsy (12,14).
The present patient had a history of recurrent
diarrhea, poor feeding and was frequently hospitalized without a
clear diagnosis. He arrived at The First Hospital of Jilin
University with reports of infection and frequent seizures. Patient
has acidosis, hepatomegaly, increase liver enzyme activity,
hyperammonemia, hypoketotic hypoglycemia and low HL activity.
Glutaric aciduria and Reye syndrome were eliminated as possible
diagnoses; Glutaric aciduria has characteristic alterations in the
GC/MS profile, including elevated concentrations of glutaryl-CoA,
glutaric acid, 3-hydroxyglutaric acid and glutarylcarnitine in body
tissues, whereas Reye syndrome shows normal GC/MS analyses
(15,16). Based on these findings, in
combination with blood and urine metabolic tests, the patient was
diagnosed with 3-HMG. No genetic testing was performed. Based on
the levels of 3-methyl-crotonyl-glycine, glutaric acid and hexane
diacid, the condition was considered to be particularly severe
(5).
During an acute 3-HMG crisis, treatment includes
glucose injection and rapid correction of acidosis, hyperammonemia,
and liver damage (17).
Administration of L-carnitine may increase the excretion of toxic
acid metabolites and prevent cardiomyopathy. Since fasting and
infection are triggers for 3-HMG onset, they should be avoided
during remission (12). For long
term maintenance, leucine-free formula and a low fat, low protein
and high carbohydrate diet is recommended (18). For patients with a family history of
3-HMG, a diagnosis can be confirmed between 13 and 23 weeks of
pregnancy by analyzing the organic acid content of the amniotic
fluid and the urine of the mother. In addition, an HL activity test
in chorionic villus cells can be performed to diagnose 3-HMG prior
to birth (5,14).
The mechanism underlying 3-HMG-associated brain
injury is unknown. Based on MRI, the most common damage is
alternation of white matter, with varying degrees of diffusion or
hypointensity in the absence or presence of atrophy (9,10). There
was one previous report of a patient with major white matter
abnormalities in the corticospinal tract on MRI (19), and another of a newborn patient with
abnormal MRI signal intensity in the thalamus and basal ganglia
(9). Fernandes et al
(20) found that intrastriatal
administration of HMG and 3-methylglutaric acid increased oxidative
stress within the rat striatum, which may explain in part the
cerebral alterations in HL deficient patients. In the present case,
MRI revealed white matter abnormality and atrophy, and the most
prominent lesion in basal ganglia was even more pronounced than
previously reported (9). The
considerable brain damage of the present patient may be due to
recurrent hypoketotic hypoglycemia, damage caused by toxic leucine
metabolites that passed through the under-developed blood-brain
barrier. In addition, the insufficient ketone body supply in this
patient may have caused myelination deficiency. Unfortunately, in
this case the parents refused further treatment, which likely
contributed to the further deterioration of the patient's
neurological condition. At the last follow-up, six month after
initial hospitalization (age, 26 months), the patient remained
unresponsive to calling and his prognosis was poor.
References
|
1
|
Aoyama Y, Yamamoto T, Sakaguchi N, Ishige
M, Tanaka T, Ichihara T, Ohara K, Kouzan H, Kinosada Y and Fukao T:
Application of multiplex ligation-dependent probe amplification,
and identification of a heterozygous Alu-associated deletion and a
uniparental disomy of chromosome 1 in two patients with
3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Int J Mol Med.
35:1554–1560. 2015.PubMed/NCBI
|
|
2
|
Faull KF, Bolton PD, Halpern B, Hammond J
and Danks DM: The urinary organic acid profile associated with
3-hydroxy-3-methylglutaric aciduria. Clin Chim Acta. 73:553–559.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ozand PT, Devol EB and Gascon GG:
Neurometabolic diseases at a national referral center: Five years
experience at the King Faisal specialist hospital and research
centre. J Child Neurol. 7(Suppl): S4–S11. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin WD, Wang CH, Lai CC, Tsai Y, Wu JY,
Chen CP and Tsai FJ: Molecular analysis of Taiwanese patients with
3-hydroxy-3-methylglutaryl CoA lyase deficiency. Clin Chim Acta.
401:33–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma YY, Song JQ, Wu TF, Liu YP, Xiao JX,
Jiang YW and Yang YL: Leucodystrophy induced by late onset
3-hydroxy-3-methylglutaric aciduria. Zhong Guo Dang Dai Er Ke Za
Zhi. 13:392–395. 2011.(In Chinese).
|
|
6
|
Leung AA, Chan AK, Ezekowitz JA and Leung
AK: A Case of Dilated Cardiomyopathy Associated with
3-Hydroxy-3-Methylglutaryl-Coenzyme A (HMG CoA) Lyase Deficiency.
Case Rep Med. 2009:1831252009.PubMed/NCBI
|
|
7
|
Puisac B, Arnedo M, Casale CH, Ribate MP,
Castiella T, Ramos FJ, Ribes A, Pérez-Cerdá C, Casals N, Hegardt FG
and Pié J: Differential HMG-CoA lyase expression in human tissues
provides clues about 3-hydroxy-3-methylglutaric aciduria. J Inherit
Metab Dis. 33:405–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hong F, Huang X, Tong F, Yang J, Yang R,
Zhou X, Huang X, Mao H and Zhao Z: A family study of
3-hydroxy-3-methylglutaric aciduria with 3 cases of sudden infant
death. Zhong Hua Er Ke Za Zhi. 52:397–399. 2014.(In Chinese).
|
|
9
|
van der Knaap MS, Bakker HD and Valk J: MR
imaging and proton spectroscopy in 3-hydroxy-3-methylglutaryl
coenzyme A lyase deficiency. AJNR Am J Neuroradiol. 19:378–382.
1998.PubMed/NCBI
|
|
10
|
Funghini S, Pasquini E, Cappellini M,
Donati MA, Morrone A, Fonda C and Zammarchi E:
3-Hydroxy-3-methylglutaric aciduria in an Italian patient is caused
by a new nonsense mutation in the HMGCL gene. Mol Genet Metab.
73:268–275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Puisac B, Ramos M, Arnedo M, Menao S,
Gil-Rodriguez MC, Teresa-Rodrigo ME, Pié A, de Karam JC, Wesselink
JJ, Giménez I, et al: Characterization of splice variants of the
genes encoding human mitochondrial HMG-CoA lyase and HMG-CoA
synthase, the main enzymes of the ketogenesis pathway. Mol Biol
Rep. 39:4777–4785. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arnedo M, Ramos M, Puisac B, Gil-Rodríguez
MC, Teresa E, Pié Á, et al: Advances in the study of genetic
disorders. Mitochondrial HMG-CoA synthase deficiency. Ikehara K:
InTech. 169–188. 2011.
|
|
13
|
Leipnitz G, Vargas CR and Wajner M:
Disturbance of redox homeostasis as a contributing underlying
pathomechanism of brain and liver alterations in
3-hydroxy-3-methylglutaryl-CoA lyase deficiency. J Inherit Metab
Dis. 38:1021–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Santarelli F, Cassanello M, Enea A, Poma
F, D'Onofrio V, Guala G, Garrone G, Puccinelli P, Caruso U, Porta F
and Spada M: A neonatal case of 3-hydroxy-3-methylglutaric-coenzyme
A lyase deficiency. Ital J Pediatr. 39:332013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boy N, Heringer J, Haege G, Glahn EM,
Hoffmann GF, Garbade SF, Kölker S and Burgard P: A cross-sectional
controlled developmental study of neuropsychological functions in
patients with glutaricaciduria type I. Orphanet J Rare Dis.
10:1632015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gallucci M, Smith JD, Limbucci N, Rossi A,
Demaerel P, Krings T, D'Amico A and Micheli C: Pediatric
Inflammatory Diseases. Part IV: Miscellaneous, Reye, PRES,
Sarcoidosis. Neuroradiol J. 25:725–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukao T, Mitchell G, Sass JO, Hori T, Orii
K and Aoyama Y: Ketone body metabolism and its defects. J Inherit
Metab Dis. 37:541–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dos Santos Mello M, Ribas GS, Wayhs CA,
Hammerschmidt T, Guerreiro GB, Favenzani JL, Sitta Â, de Moura
Coelho D, Wajner M and Vargas CR: Increased oxidative stress in
patients with 3-hydroxy-3-methylglutaric aciduria. Mol Cell
Biochem. 402:149–155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yýlmaz Y, Ozdemir N, Ekinci G, Baykal T
and Kocaman C: Corticospinal tract involvement in a patient with
3-HMG coenzyme A lyase deficiency. Pediatr Neurol. 35:139–141.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fernandes CG, da Rosa MS, Seminotti B,
Pierozan P, Martell RW, Lagranha VL, Busanello EN and Leipnitz G:
In vivo experimental evidence that the major metabolites
accumulating in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
induce oxidative stress in striatum of developing rats: A potential
pathophysiological mechanism of striatal damage in this disorder.
Mol Genet Metab. 109:144–153. 2013. View Article : Google Scholar : PubMed/NCBI
|