Introduction
Lymphangioleiomyomatosis (LAM) is a progressive
disorder that predominantly affects the lungs, and the resulting
respiratory failure can be fatal (1). The typical histology of LAM shows a
proliferation of smooth muscle arranged in fascicular, trabecular,
and papillary patterns associated with slit-like vascular channels
(2). Pulmonary LAM can occur
sporadically or in association with the inherited hamartoma
syndrome tuberous sclerosis (TSC) (3,4).
Extrapulmonary LAM is a rare disease that is usually
presented as a localized, well-circumscribed mass (5). The primary sites for LAM include the
pancreas, retroperitoneum, pelvis, mediastinum, kidney hilus,
uterus and mesentery (6). The
clinical features of extrapulmonary lymphangioleiomyomatosis are a
palpable abdominal mass, abdominal pain, and chylous ascites
(7). Differential diagnoses should
include lymphoma, schwannoma, paraganglioma at the para-aortic
area, and metastatic tumors in the lymph nodes (2). Due to the rare occurrence of
extrapulmonary LAM and its atypical location, extrapulmonary LAM is
often difficult to diagnose, especially prior to surgery. To the
best of our knowledge, the present study reports the first case of
primary LAM presenting as a liver mass.
Case report
Case summary
The present study was approved by the Ethics
Committee of the Qingdao University Medical College (Qingdao,
China). Written informed consent was obtained from the patient's
family. In 2009, a 26-year-old woman presented a liver lesion of ~6
cm diameter during a routine examination at Jiaozhou People's
Hospital of Qingdao (Qingdao, China). The liver lesion was
diagnosed as a liver cyst following a computed tomography (CT) scan
of the abdomen. The patient exhibited no clinical symptoms until
April 2012. Enhanced abdominal CT scans showed that the mass was
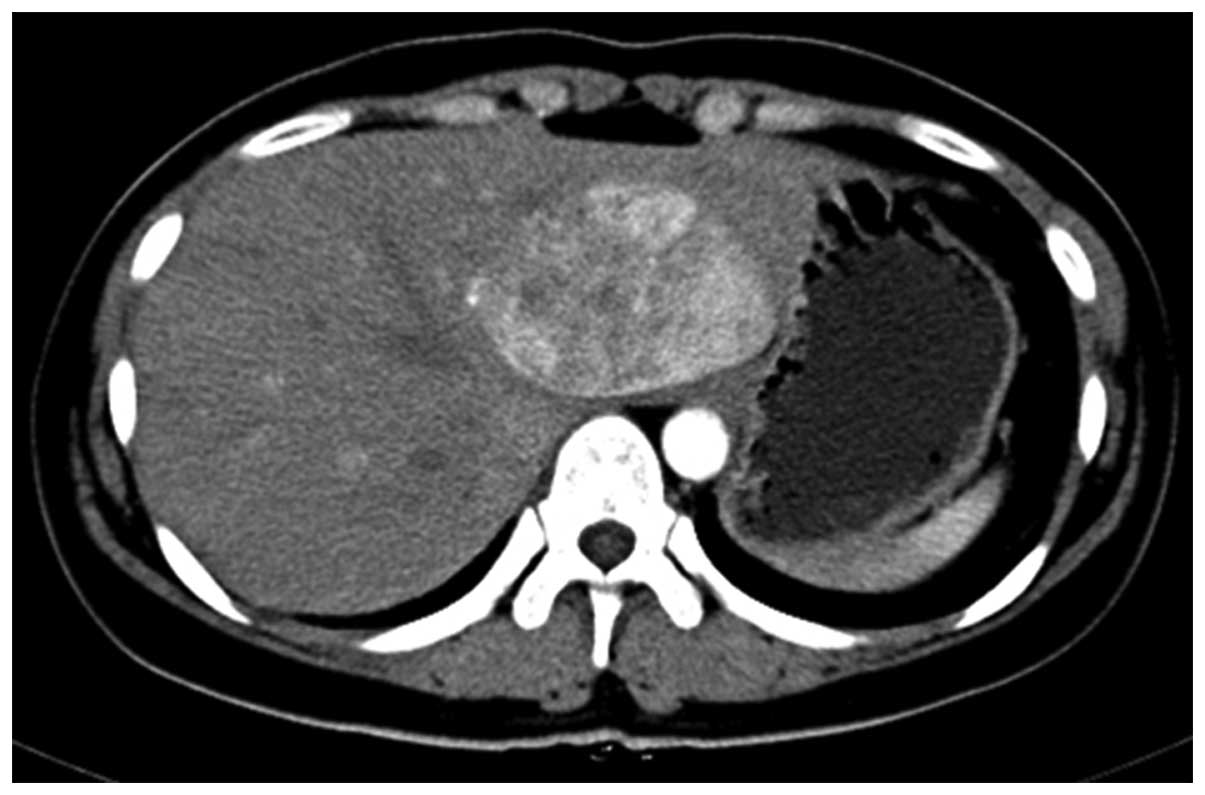
located in the left lateral hepatic lobe with 5.7×7 cm enhancement
and uneven density (Fig. 1). The
radiological differential diagnosis included left lateral hepatic
adenoma or focal nodular hyperplasia. Subsequently, a hepatic
lobectomy was performed in May, and the patient was discharged
after one week.
Pathologic findings
Gross examination demonstrated that the liver
resection specimen measured 15×10×4 cm. After the specimen was cut
longitudinally into 1-cm lobe sections, the liver lobe section
exhibited a cystic solid gray-yellow mass measuring 6×5 cm. The
size of the cysts were 0.5–2.5 cm.
Histological examination was subsequently performed.
Following fixation in 10% neutral-buffered formalin, paraffin
embedding, sectioning and staining with hematoxylin and eosin,
examination under an Olympus BX51 microscope (Olympus Corporation,
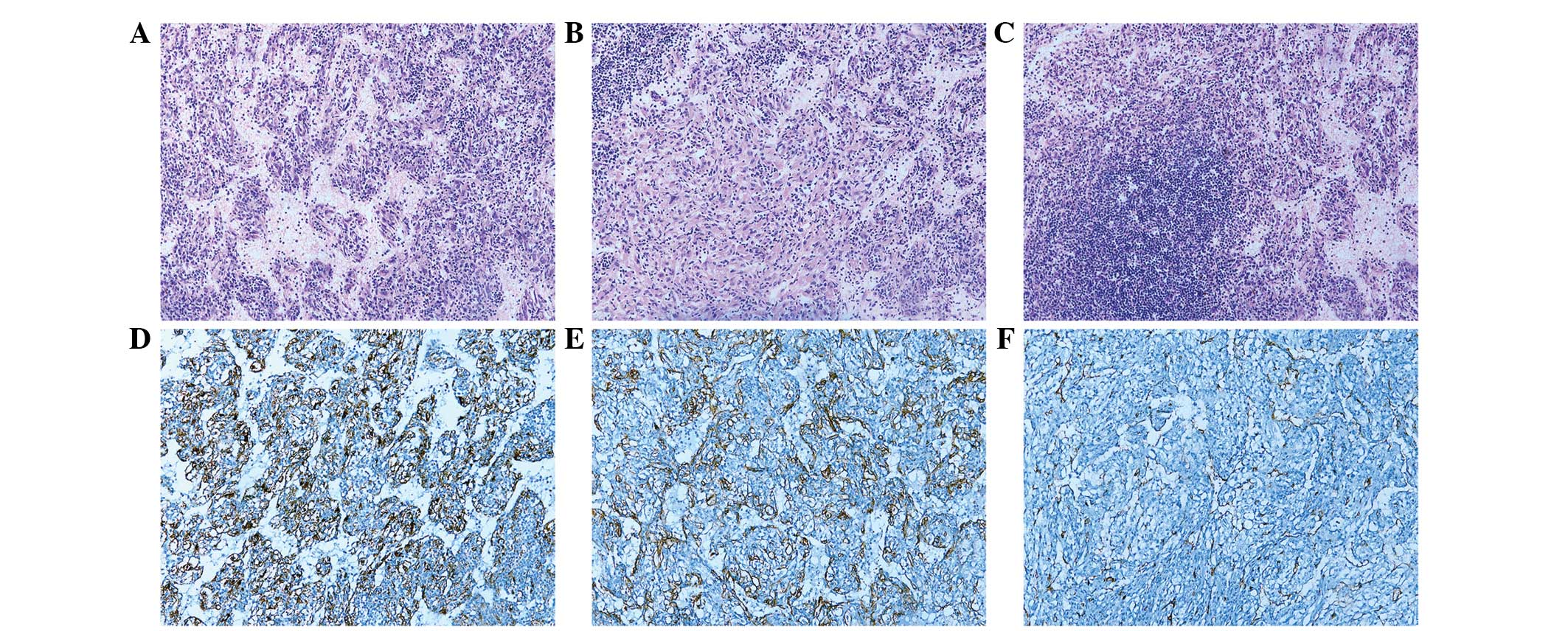
Tokyo, Japan) demonstrated that the tumor exhibited the
proliferation of spindle cells arranged along slit-like channels
lined with flattened endothelium (Fig.
2A). These spindle cells were uniform with abundant granular
eosinophilic cytoplasm and lightly stained nuclei with a smooth
nuclear membrane and inconspicuous nucleoli, which were organized
in fascicular, trabecular and anastomosing cord-like growth pattern
(Fig. 2B). In some areas, the
aggregation of lymphoid cells was observed (Fig. 2C). Images were captured using an XV
Image Processing System (Olympus Corp.).
Immunohistochemistry
Immunohistochemical staining was performed on
paraffin-embedded tissue sections (5 µm) using the EnVision method
(8). In brief, sections were
deparaffinized and pretreated in 10 mM citrate buffer solution (pH
6.0). Sections were treated with 0.3% hydrogen peroxide in methanol
for 30 min to block the endogenous peroxidase, and were
subsequently washed in phosphate-buffered saline. Sections were
incubated overnight at 4°C with: Human melanoma black-45 [working
solution (WS); HMB-45],Ki67 (1:200; K-2), Desmin (WS; D33) and
smooth muscle actin (SMA; 1:300; 1A4) mouse monoclonal antibodies;
MelanA (WS; A103) and CD31 (WS; EP78) rabbit monoclonal antibodies;
and S-100 rabbit polyclonal antibody (WS; ZA0225; all purchased
from ZSGB-BIO, Beijing, China). Following washing, section were
incubated with the secondary antibody (WS; PV6000; ZSGB-BIO) for 20
min at 37°C. A positive reaction was observed using
diaminobenzidine, followed by counterstaining with hematoxylin.
Immunohistochemistry demonstrated that the spindle cells were
positive for HMB-45 (Fig. 2D),
MelanA and SMA (Fig. 2E), but
negative for Desmin and S-100. The endothelium was positive for
CD31 (Fig. 2F). Proliferation index
with Ki-67 showed ~2% positivity in the tumor cells. The patient
did not receive any further treatment. No other lesions were
detected at biannual pulmonary and abdominal CT scans, and the
patient is well 30 months post-surgery.
Discussion
LAM is a rare neoplastic disease which predominantly
affects women with a mean age of onset of 37 years. The majority of
LAM cases occur in the lung and exhibit multiple cysts (9). Patients with pulmonary LAM present
progressive dyspnea, and pneumothorax and chylous pleural effusion.
In contrast to pulmonary LAM, extrapulmonary LAM lesions are rare
and manifest as localized lesions (10). According to previous reports, the
primary sites of extrapulmonary LAM include the posterior
mediastinum, the upper retroperitoneal areas near the abdominal
aorta and the pelvic cavity (6).
However, to the best of our knowledge, the occurrence of LAM in the
liver has yet to be reported. Only one case of abdominal LAM that
presented multiple cysts involving the liver was described by
Possekel et al (11) in a
23-year-old patient. The present case is the first extrapulmonary
LAM presenting as a liver mass without involving other organs.
LAM, together with angiomyolipoma, clear-cell
(sugar) tumor of the lung and clear-cell myomelanocytic tumor of
the falciform ligament/ligamentum teres belongs to the perivascular
epitheloid cell tumor (PEComa) family (12). PEComas has been described as ‘a
mesenchymal tumour composed of histologically and
immunohistochemically distinctive perivascular epithelioid cells’
by the World Health organization (13).
Histologically, LAM is characterized by the
proliferation of immature smooth muscle and perivascular epitheloid
cells (LAM cells) (14). These cells
are spindle-shaped or round with lightly stained nuclei with a
smooth nuclear membrane and inconspicuous nucleoli and are arranged
in a haphazard manner or in fascicular, trabecular, and papillary
patterns associated with slit-like vascular channels (2). Immunohistochemically, the tumor cells
exhibited reactivity against SMA and melanocytic (HMB-45 and
MelanA) markers (15). The present
case exhibited the distinct morphology of classical LAM which
showed the proliferation of smooth muscle spreading along the
lymphatic channels. Immunostaining revealed the tumor cells were
positive for HMB-45, MelanA and SMA. Although with distinct
morphological and immunophenotypic features extrapulmonary LAM can
be easily diagnosed, it is difficult to diagnose by radiology,
especially in the absence of accompanying pulmonary LAM.
Radiologically, the present case was initially diagnosed as hepatic
adenoma and focal nodular hyperplasia.
LAM can be divided into a sporadic form and a
tuberous sclerosis-associated form (4). Both forms can cause pulmonary symptoms.
Extrapulmonary LAM has the same morphological features as pulmonary
LAM and may occur concurrently with it or precede it (12). Matsui et al (6) described 22 cases of extrapulmonary LAM,
13 of which exhibited pulmonary LAM within 2 years. Furthermore,
3/6 cases described by Song et al (5) also exhibited concurrent pulmonary LAM
and extrapulmonary LAM. However, the present case showed a
localized liver mass without a pulmonary manifestation prior to or
following the surgical procedure, which is similar to the cases
described by Han et al (7)
and Singh et al (12).
To date, sufficient data has not been acquired
regarding the prognosis of extrapulmonary LAM. According to the
report of Matsui et al (6),
only one patient succumbed to pulmonary LAM 7 years following the
initial diagnosis, whereas the remaining 16 patients were alive at
1–13 years. The patient in the present case report is alive without
any progression 4 years following the initial surgical procedure.
As extrapulmonary LAM tends to lead to pulmonary LAM or
angiomyolipomas (6), a close
follow-up is necessary.
Studies on the therapeutic strategies for
extrapulmonary LAM have been limited. Current studies provide the
foundation for novel therapeutic strategies for LAM. LAM occurs in
~30% of woman with TSC (16–18), and has therefore been well-described
as a complication of tuberous sclerosis associated with mutations
in the tuberous sclerosis genes TSC1 and TSC2 (19). Specifically, TSC genes (TSC1 and
TSC2) have an important role in the regulation of the cell cycle
via the mammalian target of rapamycin (mTOR) signaling pathway
(20). Sirolimus, also called
rapamycin (an mTOR inhibitor) blocks the mTOR signaling pathway and
restores homeostasis in the LAM cell (18). Possekel et al (11) described a 23-year-old woman with
abdominal manifestations of LAM, who received sirolimus therapy and
showed clinical benefit. On the other hand, anti-estrogenic
therapy, such as oophorectomy and progesterone, has also been
demonstrated to be an alternative way to treat LAM (21). Application of hormone therapy in
extrapulmonary LAM had been reported, whereas the guidelines of the
European Respiratory Society on the diagnosis and treatment of LAM
(16) published in 2010, did not
recommend the standard use of progesterone.
To conclude, the present study presents a case of
localized liver LAM with fibrous capsule in a 26-year-old woman
with no symptoms. Radiological examination revealed a potential
hepatic adenoma or focal nodular hyperplasia. However, pathological
examination revealed the liver lesion to be a histologically and
immunohistochemically classic LAM. Therefore, for female patients
in whom liver masses are observed, extrapulmonary LAM should be
considered in the differential diagnosis.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant no. 81201947).
References
|
1
|
Li C, Lee PS, Sun Y, Gu X, Zhang E, Guo Y,
Wu CL, Auricchio N, Priolo C, Li J, et al: Estradiol and mTORC2
cooperate to enhance prostaglandin biosynthesis and tumorigenesis
in TSC2-deficient LAM cells. J Exp Med. 211:15–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaiswal VR, Baird J, Fleming J, Miller DS,
Sharma S and Molberg K: Localized retroperitoneal
lymphangioleiomyomatosis mimicking malignancy. A case report and
review of the literature. Arch Pathol Lab Med. 127:879–882.
2003.PubMed/NCBI
|
|
3
|
Johnson SR: Lymphangioleiomyomatosis. Eur
Respir J. 27:1056–1065. 2006.PubMed/NCBI
|
|
4
|
Taveira-DaSilva AM, Pacheco-Rodriguez G
and Moss J: The natural history of lymphangioleiomyomatosis:
Markers of severity, rate of progression and prognosis. Lymphatic
Res Biol. 8:9–19. 2010. View Article : Google Scholar
|
|
5
|
Song DH, Choi IH, Ha SY, Han KM, Lee JJ,
Hong ME, Choi YL, Jang KT, Song SY, Yi CA and Han J: Extrapulmonary
lymphangioleiomyoma: Clinicopathological analysis of 4 cases.
Korean J Pathol. 48:188–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matsui K, Tatsuguchi A, Valencia J, Yu Zx,
Bechtle J, Beasley MB, Avila N, Travis WD, Moss J and Ferrans VJ:
Extrapulmonary lymphangioleiomyomatosis (LAM): Clinicopathologic
features in 22 cases. Hum Pathol. 31:1242–1248. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han JM, Lee KH, Kim SJ, Rhim CC, Park YH,
Kang JB and Jeon SY: A case of lymphangioleiomyomatosis originated
in the pelvic cavity. J Gynecol Oncol. 19:195–198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wenjuan Y, Yujun L and Ceng Y: Association
of single nucleotide polymorphisms of beta2-adrenergic receptor
gene with clinicopathological features of pancreatic carcinoma.
Acta Histochem. 115:198–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu SH, Hou YY, Tan YS, Xu JF, Zeng HY,
Sujie AK, Wang XD and Bai CX: Clinical and histopathological
alterations of lymphangioleiomyomatosis in 14 Chinese patients.
Chinese Med J. 122:1895–1900. 2009.
|
|
10
|
Kitaichi M, Nishimura K, Itoh H and Izumi
T: Pulmonary lymphangioleiomyomatosis: A report of 46 patients
including a clinicopathologic study of prognostic factors. Am J
Resp Crit Care Med. 151:527–533. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Possekel AK, Katenkamp D, Brambs HJ and
Pauls S: Lymphangioleiomyomatosis: Solitary abdominal manifestation
(2009: 9b). Eur Radiol. 19:3015–3018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh M, Saroha V, Wadhwa R, Khurana N and
Kakkar AK: Solitary lymphangioleiomyoma of pancreas mimicking
pancreatic pseudocyst-a case report. J Gastrointest Cancer.
43:336–339. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foipe AL: Neoplasms with perivascular
epithelioid cell differentiation. World Health organization
classification of tumours: Pathology and genetics of tumours of
soft tissue and bone. Fletcher CD, Unni KK, Mertens F, et al: LARC
Press. (Lyon). 221–222. 2002.
|
|
14
|
Grzegorek I, Drozdz K, Podhorska-Okolow M,
Szuba A and Dziegiel P: LAM cells biology and
lymphangioleiomyomatosis. Folia Histochem Cytobiol. 51:1–10. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hornick JL and Fletcher CD: PEComa: What
do we know so far? Histopathology. 48:75–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson SR, Cordier JF, Lazor R, Cottin V,
Costabel U, Harari S, Reynaud-Gaubert M, Boehler A, Brauner M,
Popper H, et al: European Respiratory Society guidelines for the
diagnosis and management of lymphangioleiomyomatosis. Eur Respir J.
35:14–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Casanova A and Ancochea J:
Lymphangioleiomyomatosis: new therapeutic approaches. Arch
Bronconeumol. 47:579–580. 2011.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derweduwen AM, Verbeken E, Stas M,
Verschakelen J, Coolen J, Verleden G and Wuyts W: Extrapulmonary
lymphangioleiomyomatosis: A wolf in sheep's clothing. Thorax.
68:111–113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCormack FX: Lymphangioleiomyomatosis: A
clinical update. Chest. 133:507–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goncharova EA, Goncharov DA, Eszterhas A,
Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski
DJ, Chou MM, et al: Tuberin regulates p70 S6 kinase activation and
ribosomal protein S6 phosphorylation. A role for the TSC2 tumor
suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol
Chem. 277:30958–30967. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ohori NP, Yousem SA, Sonmez-Alpan E and
Colby TV: Estrogen and progesterone receptors in
lymphangioleiomyomatosis, epithelioid hemangioendothelioma and
sclerosing hemangioma of the lung. Am J Clin Pathol. 96:529–535.
1991. View Article : Google Scholar : PubMed/NCBI
|