Introduction
Myocardial infarction (MI) is the most common cause
of mortality and disability, with a direct correlation between
infarct size and prognosis (1).
Reperfusion of ischemic tissue is necessary to terminate the
processes of ischemic injury that ultimately results in infarction.
However, abrupt reperfusion may be associated with severe metabolic
and ionic disturbances that can provoke further tissue injury and
myocardial cell death [ischemia/reperfusion (I/R) injury] (2). Since reperfusion is the cornerstone of
treatment for acute MI, there is great interest in the development
of adjunct therapies, which may attenuate reperfusion injury and
thereby maximize the benefits of reperfusion (1,3).
Adenosine and selective adenosine receptor agonists have been
studied extensively for their ability to reduce infarct size and
apoptosis (4–8). These effects appear to be mediated via
the activation of one or more adenosine receptor subtypes (7). Adenosine-5′-triphosphate (ATP) is
rapidly converted to adenosine by ectonucleotidases, and coronary
vasodilation caused by exogenous ATP is entirely mediated by
adenosine acting on A2A-adenosine receptors (9). We previously demonstrated that
cardioprotection of ischemic postconditioning and
ATP-postconditioning in rabbits is associated with the activation
of adenosine receptors (10).
Previous findings showed that ischemic
postconditioning exerts protective effects through the recruitment
of prosurvival kinases such as phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (PKB)/Akt and the p42/p44 extracellular
signal-regulated kinase 1/2 (ERK1/2) pathways [also known as
reperfusion injury salvage kinase (RISK) pathway] at the time of
reperfusion (11,12). However, whether exogenous ATP can
induce postconditioning effects in the myocardium or whether these
effects are mediated through a mechanism similar to that of
ischemic postconditioning has yet to be fully examined.
Therefore, in the present study, using an in
vivo rabbit model of acute MI, we examined the acute effects of
ATP on myocardial infarct size and apoptosis inhibition as well as
its precise molecular mechanism involved in the activation of
specific survival signals (PI3K/AKT and ERK1/2 pathways).
Materials and methods
Experimental animals
Sixty male New Zealand white rabbits with a body
weight of 2.0–2.5 kg, were used in the present study. The rabbits
were housed in a temperature-controlled environment (21±2°C) on a
12-h light/dark cycle (lights on at 06:00). The animals had free
access to food and water. Facilities housing the animals were
followed guidelines of the AAALAC (the Association for Assessment
and Accreditation of Laboratory Animal Care International),
accredited at the time of the study. The study protocol was
approved by the Ethics Committee of Qingdao University School of
Medicine (Qingdao, China).
Reagents
Wortmannin (PI3K inhibitor), PD-98059 (ERK
inhibitor), and 5-hydroxydecanoic acid (5-HD) (mitochondrial
ATP-dependent potassium ion channel blocker) were purchased from
Sigma Chemical Co. (St. Louis, MO, USA). ATP was purchased from the
Tianjin Pharmaceutical Group Jiao-Zuo Co. (Tianjin, China). To
detect and quantify apoptosis, a terminal
deoxynucleotidyl-transferase-mediated dUTP nick end-labeling
(TUNEL) assay was performed according to the manufacturer's
instructions using a commercially available kit (Roche, Basel,
Switzerland). Any other reagents used were of standard analytical
grade.
Surgical preparation
Male New Zealand white rabbits were anesthetized
with urethane (5 ml/kg). Surgical procedures were performed
aseptically. The left carotid artery was cannulated to monitor
arterial pressure, and electrocardiogram (ECG) leads were placed to
record the heart rate. A polyethylene catheter (0.9-mm lumen
diameter) was inserted into the internal carotid artery and was
advanced 1 cm towards the heart to monitor blood pressure. Blood
pressure was measured using a fluid-filled pressure transducer
connected to the end of the cannula. Arterial blood pressure and
the heart rate were measured via a catheter introduced into the
carotid artery. A micromanometer-tipped catheter (SPR-407; Millar
Instruments, Houston, TX, USA) was inserted into the left ventricle
to record +dp/dtmax (representing the cardiac systolic
function) as well as −dp/dtmax (representing cardiac
diastolic function). Drugs and saline were administered via the ear
vein. After left thoracotomy was performed in the third and fourth
intercostal spaces in the exposed heart, a 4/0 silk thread was
placed beneath the large arterial branch coursing down the middle
of the anterolateral surface of the left ventricle. Coronary
arterial occlusion and reperfusion were performed by pushing or
releasing the snare made from thread. A prominent anterior branch
of the left coronary artery was under-run with a 3/0 silk suture,
the ends of which were threaded through a 13-mm polypropylene tube
to form a snare. After the administration of heparin sodium at a
dose of 300 IU/kg−1, regional myocardial ischemia was
induced by clamping the snare with hemostat forceps. Reperfusion
was instituted by releasing the snare. Coronary arterial occlusion
was confirmed by observing cyanosis of the myocardium as well as
ST-segment deviation.
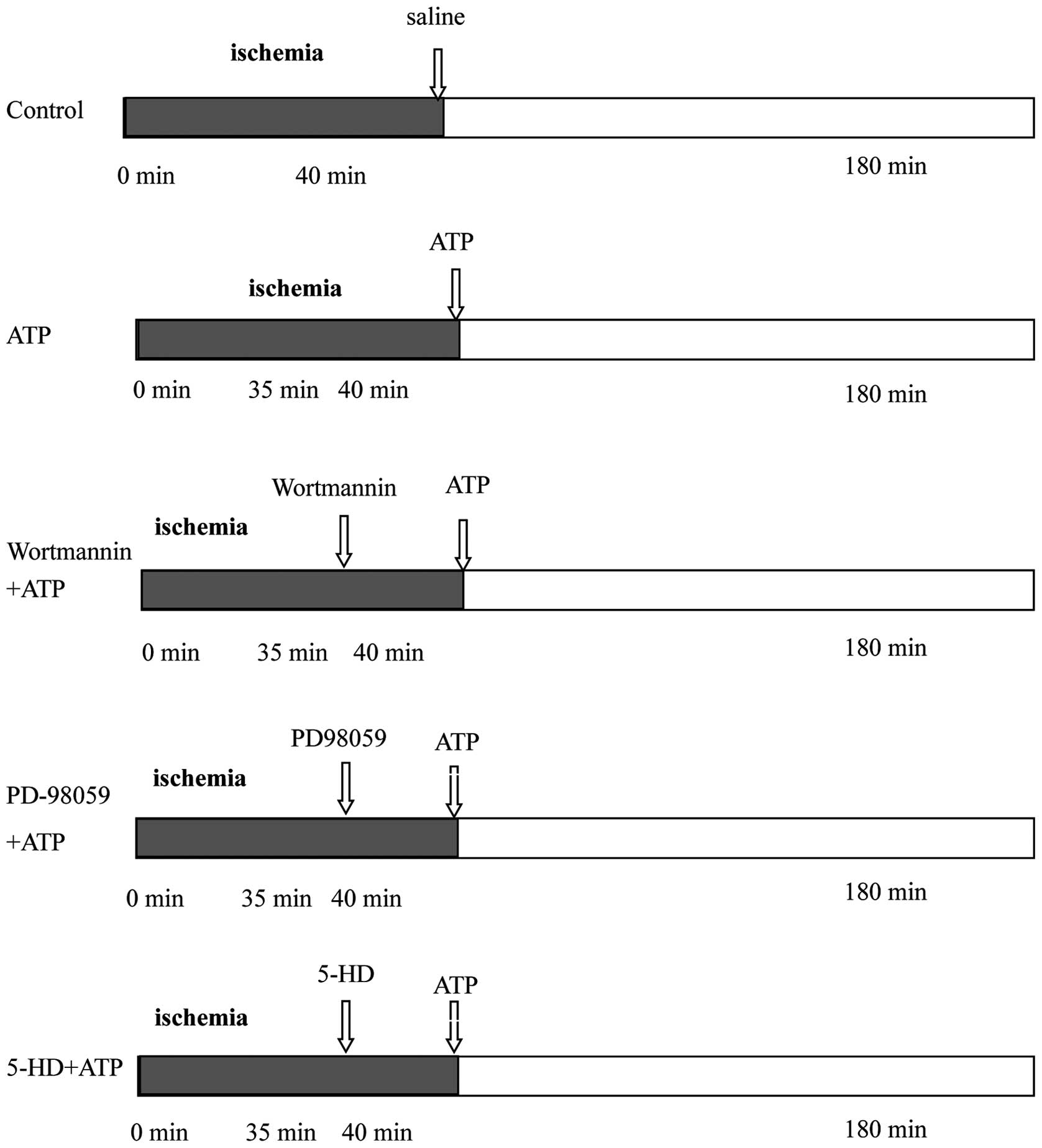
Sixty male New Zealand white rabbits underwent 40
min of coronary occlusion followed by 180 min of reperfusion, and
were then assigned randomly to 5 groups (n=12 for each group)
(Fig. 1). For the control group,
0.9% NaCl was administered intravenously immediately after
reperfusion and maintained throughout the first 30 min. The ATP
group was identical to the control group except that ATP (3 mg/kg)
was administered intravenously and maintained throughout the first
30 min instead of saline. The wortmannin+ATP, PD-98059+ATP, and
5-HD+ATP groups were identical to the ATP group except that
wortmannin (PI3K inhibitor, 0.6 mg/kg), PD-98059 (ERK inhibitor,
0.3 mg/kg), or 5-HD [mitochondrial ATP-sensitive K+
(mitoKATP) channel blocker, 5 mg/kg] were injected
intravenously as a bolus, 5 min prior to initiation of ATP
injection in the respective groups.
Analysis of MI size
After the 3-h reperfusion period, the coronary
branch was reoccluded and Evans Blue dye solution (4 ml, 2% w/v)
was injected into the left ventricle to distinguish between
perfused and non-perfused (myocardium at risk) sections of the
heart. The Evans Blue solution stained the perfused myocardium,
while the occluded vascular bed was not stained. The rabbits were
sacrificed using an intravenous overdose of pentobarbital (100–200
mg/kg). The heart was excised and sectioned into 4- to 5-µm thick
sections. After removing the right ventricular wall, the area at
risk and non-ischemic myocardium were separated by following the
line of demarcation between blue-stained and unstained (pink/red)
tissue. To distinguish between ischemic and infarcted tissue, the
area at risk was cut into small sections and incubated (20 min at
37°C) with p-nitro-blue tetrazolium (NBT, 0.5 mg ml-1; Sigma
Chemical Co.). In the presence of intact dehydrogenase enzyme
systems (normal myocardium), NBT forms a dark blue formazan, while
areas of necrosis lack dehydrogenase activity and therefore showed
no staining. The area at risk (AAR, area without blue dye) was
identified and traced from the enlarged projection (×10) of the
photographic slide of each ventricular slice. AAR and IS were
determined by computerized planimetry using ImageJ software
(Chicago, IL, USA). AAR was expressed as a percentage of the left
ventricle and IS was expressed as a percentage of the AAR.
Detection of apoptosis
The detection of apoptotic cells was performed using
TUNEL as previously reported (13).
The tissue blocks were fixed in 4% paraformaldehyde and incubated
with proteinase K. Fragments of DNA in the tissue sections were
analyzed using a TUNEL detection kit (Roche). For each slide, the
color images of 10 separate fields were captured randomly and
digitized. The cells with clear nuclear labeling were defined as
TUNEL-positive cells. The apoptotic index (AI) was calculated as
the number of TUNEL-positive cells/total number of myocytes ×
100.
Western blot analysis
Western blot analysis was performed to assess the
levels of Akt and p-Akt, as well as ERK and p-ERK in the ischemic
area of the myocardium following 180-min reperfusion. Hearts were
excised, and transmural samples weighing 100–200 mg were obtained
from the center of the left ventricular (LV) ischemic region. The
border of the ischemic region was defined by the distribution of
cyanosis and was marked on the epicardium in ink. Tissue samples
obtained after perfusion were quickly frozen in liquid nitrogen and
stored at −80°C until the assays were performed. The samples were
weighed, homogenized, and used for different measurements. Proteins
were separated and transferred to membranes using standard
protocols, after which the phosphorylation (activation) of Akt and
ERK and total levels of Akt and ERK were assessed using antibodies
against each protein, and then analyzed by SDS-PAGE
immunoelectrophoresis.
Statistical analysis
Data were expressed as means ± SE and analyzed using
SPSS 17.0 software (Chicago, IL, USA). Independent samples t-test
and one-way ANOVA were used to compare data with post-hoc analysis
using Bonferroni's post-hoc test. Differences at p<0.05 were
considered statistically significant.
Results
Physiological findings
Table I shows the
hemodynamic parameters that may influence the infarct size. There
were no significant differences in blood pressure or heart rate in
the five groups at 180 min after reperfusion. However, 180 min
after reperfusion, ±dp/dt was significantly improved in the ATP
group as compared to the remaining four groups.
 | Table I.Hemodynamic parameters. |
Table I.
Hemodynamic parameters.
| Group | Heart rate
(beats/min) | Mean blood pressure
(mmHg) | +dp/dt
(mmHg/sec) | −dp/dt
(mmHg/sec) |
|---|
| Control |
238.8±7.28 |
72.5±4.23 |
2924.3±157.69 |
2325.00±374.60 |
| ATP |
240.2±6.65 |
71.0±5.18 |
4432.17±221.78a |
4129±136.90b |
| Wortmannin+ATP |
240.0±6.13 |
72.0±6.9 |
2872.80±152.6 |
2162.00±270.3 |
| PD-98059+ATP |
243.2±7.46 |
70.5±6.9 |
2753.8±178.25 |
2074.67±279.65 |
| 5-HD+ATP |
237.8±7.08 |
72.2±5.98 |
2783.5±128.98 |
2206.33±197.49 |
Infarct size
As shown in Table
II, the percentage of infarct size in the area at risk was
significantly reduced in the ATP group (12.79±1.87%, n=6), as
compared with the saline control group (29.10±2.94%, n=6). However,
pretreatment with wortmannin (n=6), PD-98059 (n=6), or 5-HD (n=6)
completely eliminated the infarct size-reducing effect of ATP
(26.54±2.71, 27.93±3.18 and 26.04±4.03%, respectively).
| Table II.Comparison of infarct size and AI at
the end of reperfusion. |
Table II.
Comparison of infarct size and AI at
the end of reperfusion.
| Group | Infarct size (%) | AI (%) |
|---|
| Control |
29.10±2.94 |
27.00±5.76 |
| ATP |
12.79±1.87a |
10.33±5.96b |
| Wortmannin+ATP |
26.54±2.71 |
20.67±4.32 |
| PD-98059+ATP |
27.93±3.18 |
25.50±4.85 |
| 5-HD+ATP |
26.04±4.03 |
21.17±3.60 |
Myocardial apoptosis after
reperfusion
There was increased apoptosis 180 min after
reperfusion in all the groups, except for the ATP group (Table II), indicating that ATP
significantly reduced myocardial apoptosis during early
reperfusion. Treatment with wortmannin, PD98059, or 5-HD
significantly attenuated the anti-apoptotic effects of ATP.
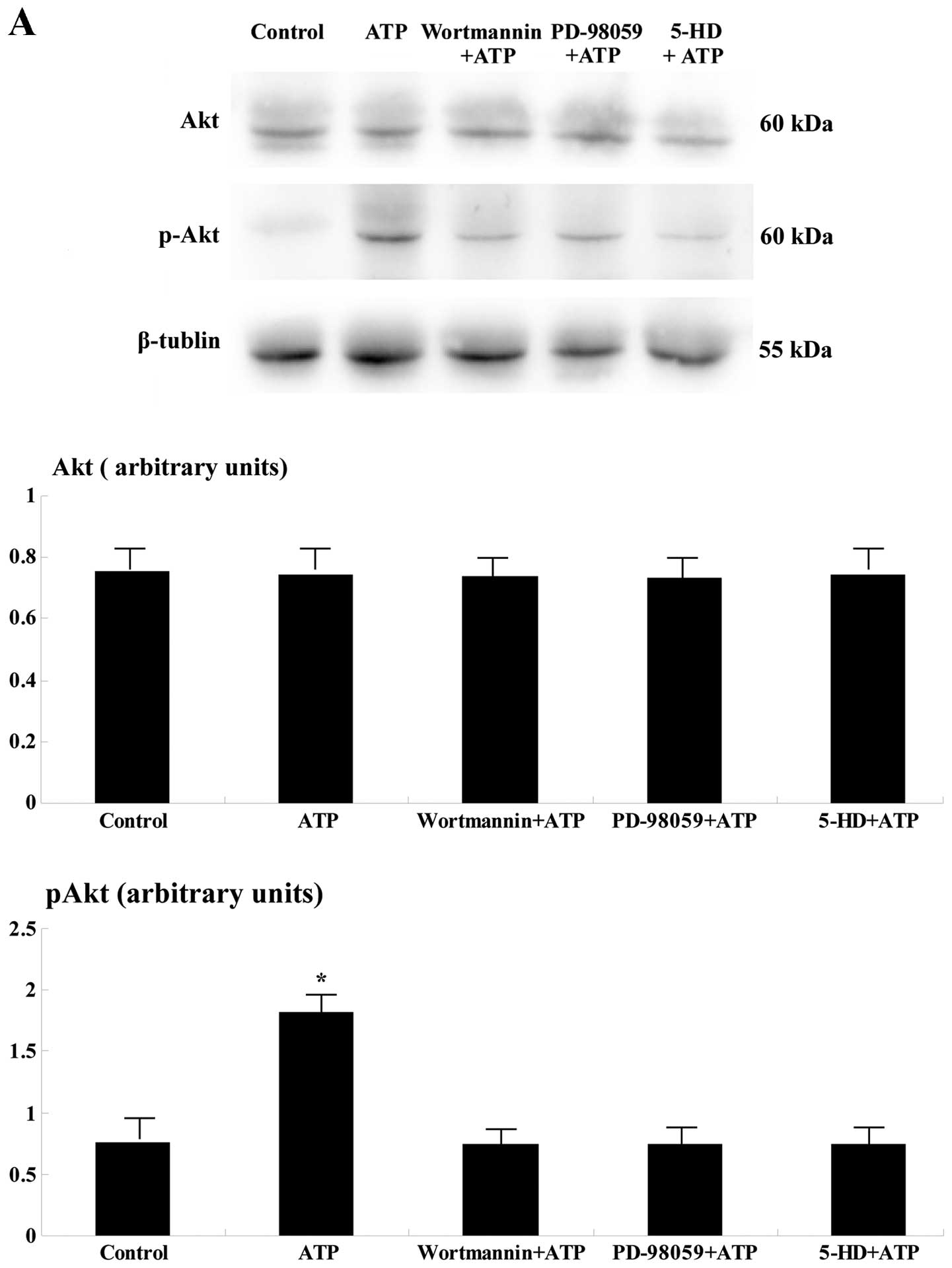
Western blot analysis
One hundred eighty minutes after reperfusion, the
levels of p-Akt and p-ERK were significantly upregulated in the
ischemic area of the ATP group when compared to the remaining four
groups (p<0.05; Fig. 2).
Discussion
ATP and adenosine are potent coronary vasodilators.
Similarly, exogenous ATP is almost completely metabolized to
adenosine during a single passage through the heart (14). In humans, the intracoronary
administration of ATP causes vasodilation, which is accompanied by
an increase in the concentration of coronary sinus adenosine
(15). Thus, ATP, as well as
adenosine, may be used as cardioprotective agents.
MI is typically associated with apoptosis and the
progressive loss of cardiomyocytes caused by apoptosis plays a
critical role in cardiac dysfunction after acute MI (16). The present study clearly demonstrates
that the intravenous administration of ATP may significantly
attenuate cardiomyocyte apoptosis and reduce infarct size. The
underlying mechanism may be associated with the inhibition of
oxidation stress, upregulation of Bcl-2-encoding mRNA and
downregulation of caspase-3 mRNA. Using a swine I/R model Vilahur
et al have demonstrated that ischemic postconditioning
prevents execution of apoptosis (via Bcl-2 and caspase-3),
supporting our observation of the effect induced by ATP
administration (17).
Activation of the PI3K/Akt and MEK1/2-ERK1/2
pathways is involved in the infarct size-reducing effect during
ischemic postconditioning, as prosurvival signals (18–20). Our
observations suggest that ATP treatment during reperfusion
activates the PI3K/Akt and MEK1/2-ERK1/2 pathways in the ischemic
area. Therefore, since ATP treatment during reperfusion attenuates
cardiomyocyte apoptosis and reduces infarct size through activation
of the prosurvival signaling pathways such as PI3K/Akt and
MEK1/2-ERK1/2, the infarct size-reducing effects of ATP were
mediated through a mechanism similar to that of ischemic
postconditioning. It has been reported that the adenosine A1/A2
agonist, 5′-N-ethylcarboxamidoadenosine (NECA) and bradykinin can
limit infarction when administered at reperfusion in rabbits
through a common signaling pathway that includes PI3K, nitric oxide
(NO), and ERK (21). The infarct
size-reducing effect of ATP is suggested to be due to the
activation of PI3K and Akt and subsequent phosphorylation of
endothelial nitric oxide synthase (eNOS). Cohen et al
previously found that the protective effect of protein kinase G
(PKG) activator during reperfusion can be blocked by A2b adenosine
receptor, ERK, or PI3K blockers (22).
Concerning the mitoKATP channels, it has
been reported that either ischemic preconditioning or
postconditioning is an effective cardioprotective intervention in
rabbits, involving a protective mechanism of NO production as well
as mitoKATP channel opening (23,24). In
the present study, the infarct size-reducing effect of ATP was
eliminated by pretreatment with 5-HD, a mitoKATP channel
blocker, suggesting that the infarct-reducing effect by ATP was
mediated by opening the mitoKATP channels. Since NO has
been reported to open mitoKATP channels (25), it is possible that ATP activates the
PI3K/Akt and eNOS pathways, opens mitoKATP channels, and
reduces myocardial infarct size. Furthermore, administration of ATP
may increase NO production in the heart (26) and reduce the extent of no-reflow and
infarct size (27). As NO and
adenosine are important in cardioprotection, the administration of
ATP may induce postconditioning in myocardium.
In conclusion, our findings demonstrate that ATP
administration immediately after reperfusion reduces myocardial
infarct size and exerts a significant cardioprotective effect
against I/R injury by inhibiting apoptosis and improving LV
function. Cardioprotection by postischemic ATP administration is
mediated through activation of the reperfusion injury salvage
kinase pathway and opening of the mitoKATP channels.
Clinical implications
Pharmacological postconditioning is a more practical
strategy than preconditioning for treating MI, because it is
difficult to predict the timing precisely when acute MI occurs.
Previously, however, no pharmacologic adjunctive therapy in
patients with acute MI undergoing reperfusion therapy was clearly
demonstrated to reduce infarct size and improve clinical outcomes
except for adenosine (28–30). Furthermore, the intravenous infusion
of ATP may have fewer side effects and be safer than adenosine
(31). Therefore, in the clinical
setting, pharmacological postconditioning may be induced with
exogenous ATP as shown in the present study and may be an
alternative strategy for the treatment of acute MI during
reperfusion, although further clinical investigations are
necessary.
Limitations and future investigations
The major limitation of the present study was that
our results did not reveal the protective effect of ATP on
preventing myocardial apoptosis during prolonged reperfusion and
its optimal timing and dosing. Furthermore, different mechanisms of
action for adenosine and ATP may exist. A parallel experimental
approach based on the administration of adenosine and adenosine
receptor inhibitors may have been of interest to reveal potential
differences in the cardioprotective mechanisms.
In conclusion, cardioprotection by postischemic ATP
administration is mediated through activation of the reperfusion
injury salvage kinase (RISK) pathway and opening of the
mitochondrial ATP-dependent potassium channels. However, the
results of the present study remain to be verified in future
investigations.
Acknowledgements
We would like to thank Mrs. Nini Gao for her expert
editorial assistance and help with manuscript preparation. The
present study was supported by the Youth Research Development Funds
of the Affiliated Hospital of Qingdao University (200804).
References
|
1
|
Yellon DM and Baxter GF: Protecting the
ischaemic and reperfused myocardium in acute myocardial infarction:
distant dream or near reality? Heart. 83:381–387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hearse DJ and Bolli R: Reperfusion induced
injury: manifestations, mechanisms, and clinical relevance.
Cardiovasc Res. 26:101–108. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yellon DM and Baxter GF: Reperfusion
injury revisited: is there a role for growth factor signaling in
limiting lethal reperfusion injury? Trends Cardiovasc Med.
9:245–249. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuno A, Solenkova NV, Solodushko V, Dost
T, Liu Y, Yang XM, Cohen MV and Downey JM: Infarct limitation by a
protein kinase G activator at reperfusion in rabbit hearts is
dependent on sensitizing the heart to A2b agonists by protein
kinase C. Am J Physiol Heart Circ Physiol. 295:H1288–H1295. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Urmaliya VB, Pouton CW, Ledent C, Short JL
and White PJ: Cooperative cardioprotection through adenosine A1 and
A2A receptor agonism in ischemia-reperfused isolated mouse heart. J
Cardiovasc Pharmacol. 56:379–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kin H, Zatta AJ, Lofye MT, Amerson BS,
Halkos ME, Kerendi F, Zhao ZQ, Guyton RA, Headrick JP and
Vinten-Johansen J: Postconditioning reduces infarct size via
adenosine receptor activation by endogenous adenosine. Cardiovasc
Res. 67:124–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Regan SE, Broad M, Byford AM, Lankford AR,
Cerniway RJ, Mayo MW and Matherne GP: A1 adenosine receptor
overexpression attenuates ischemia-reperfusion-induced apoptosis
and caspase 3 activity. Am J Physiol Heart Circ Physiol.
284:H859–H866. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Philipp S, Yang XM, Cui L, Davis AM,
Downey JM and Cohen MV: Postconditioning protects rabbit hearts
through a protein kinase C-adenosine A2b receptor cascade.
Cardiovasc Res. 70:308–314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Erga KS, Seubert CN, Liang HX, Wu L,
Shryock JC and Belardinelli L: Role of A(2A)-adenosine receptor
activation for ATP-mediated coronary vasodilation in guinea-pig
isolated heart. Br J Pharmacol. 130:1065–1075. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lian ZX, Liu F, Liu S, Xin H, Chen ZY,
Tian JH, An Y and Cai SL: Cardioprotection of ischemic
postconditioning and ATP-postconditioning in rabbits is associated
with the activation of adenosine receptors. Eur Heart J. 27:(Suppl
1). 722006.
|
|
11
|
Hausenloy DJ, Tsang A and Yellon DM: The
reperfusion injury salvage kinase pathway: a common target for both
ischemic preconditioning and postconditioning. Trends Cardiovasc
Med. 15:69–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bopassa J-C, Ferrera R, Gateau-Roesch O,
Couture-Lepetit E and Ovize M: PI 3-kinase regulates the
mitochondrial transition pore in controlled reperfusion and
postconditioning. Cardiovasc Res. 69:178–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kin H, Wang NP, Mykytenko J, Reeves J,
Deneve J, Jiang R, Zatta AJ, Guyton RA, Vinten-Johansen J and Zhao
ZQ: Inhibition of myocardial apoptosis by postconditioning is
associated with attenuation of oxidative stress-mediated nuclear
factor-kappa B translocation and TNF alpha release. Shock.
29:761–768. 2008.PubMed/NCBI
|
|
14
|
Fleetwood G, Coade SB, Gordon JL and
Pearson JD: Kinetics of adenine nucleotide catabolism in coronary
circulation of rats. Am J Physiol. 256:H1565–H1572. 1989.PubMed/NCBI
|
|
15
|
Nanto S, Kitakaze M, Takano Y, Hori M and
Nagata S: Intracoronary administration of adenosine triphosphate
increases myocardial adenosine levels and coronary blood flow in
man. Jpn Circ J. 61:836–842. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Olivetti G, Quaini F, Sala R, Lagrasta C,
Corradi D, Bonacina E, Gambert SR, Cigola E and Anversa P: Acute
myocardial infarction in humans is associated with activation of
programmed myocyte cell death in the surviving portion of the
heart. J Mol Cell Cardiol. 28:2005–2016. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vilahur G, Cubedo J, Casani L, Padro T,
Sabate-Tenas M, Badimon JJ and Badimon L: Reperfusion-triggered
stress protein response in the myocardium is blocked by
post-conditioning. Systems biology pathway analysis highlights the
key role of the canonical aryl-hydrocarbon receptor pathway. Eur
Heart J. 34:2082–2093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heusch G, Boengler K and Schulz R:
Cardioprotection: nitric oxide, protein kinases, and mitochondria.
Circulation. 118:1915–1919. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goodman MD, Koch SE, Fuller-Bicer GA and
Butler KL: Regulating RISK: a role for JAK-STAT signaling in
postconditioning? Am J Physiol Heart Circ Physiol. 295:H1649–H1656.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu M, Feng J, Lucchinetti E, Fischer G,
Xu L, Pedrazzini T, Schaub MC and Zaugg M: Ischemic
postconditioning protects remodeled myocardium via the PI3K-PKB/Akt
reperfusion injury salvage kinase pathway. Cardiovasc Res.
72:152–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang XM, Krieg T, Cui L, Downey JM and
Cohen MV: NECA and bradykinin at reperfusion reduce infarction in
rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell
Cardiol. 36:411–421. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cohen MV, Yang XM, Liu Y, Solenkova NV and
Downey JM: Cardioprotective PKG-independent NO signaling at
reperfusion. Am J Physiol Heart Circ Physiol. 299:H2028–H2036.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hide EJ and Thiemermann C: Limitation of
myocardial infarct size in the rabbit by ischaemic preconditioning
is abolished by sodium 5-hydroxydecanoate. Cardiovasc Res.
31:941–946. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang XM, Proctor JB, Cui L, Krieg T,
Downey JM and Cohen MV: Multiple, brief coronary occlusions during
early reperfusion protect rabbit hearts by targeting cell signaling
pathways. J Am Coll Cardiol. 44:1103–1110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han J, Kim N, Joo H, Kim E and Earm YE:
ATP-sensitive K(+) channel activation by nitric oxide and protein
kinase G in rabbit ventricular myocytes. Am J Physiol Heart Circ
Physiol. 283:H1545–H1554. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burnstock G: Overview: purinergic
receptorsRole of adenosine and adenosine nucleotides in the
biological system. Imai S and Nakazawa M: Elsevier Science;
Amsterdam: pp. 1–6. 1991
|
|
27
|
Komamura K, Ito H, Takiuchi S, Iwakura K,
Maruyama A, Masuyama T, Minamino T, Node K, Kitakaze M and Hori M:
Transient intracoronary infusion of ATP after reperfusion reduces
the extent of no-reflow and infarct size in dogs. J Am Coll
Cardiol. 25:227A–228A. 1995. View Article : Google Scholar
|
|
28
|
Mahaffey KW, Puma JA, Barbagelata NA,
DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC,
Molina-Viamonte V, et al: Adenosine as an adjunct to thrombolytic
therapy for acute myocardial infarction: results of a multicenter,
randomized, placebo-controlled trial: the Acute Myocardial
Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol.
34:1711–1720. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ross AM, Gibbons RJ, Stone GW, Kloner RA
and Alexander RW: AMISTAD-II Investigators: A randomized,
double-blinded, placebo-controlled multicenter trial of adenosine
as an adjunct to reperfusion in the treatment of acute myocardial
infarction (AMISTAD-II). J Am Coll Cardiol. 45:1775–1780. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kloner RA, Forman MB, Gibbons RJ, Ross AM,
Alexander RW and Stone GW: Impact of time to therapy and
reperfusion modality on the efficacy of adenosine in acute
myocardial infarction: the AMISTAD-2 trial. Eur Heart J.
27:2400–2405. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miyagawa M, Kumano S, Sekiya M, Watanabe
K, Akutzu H, Imachi T, Tanada S and Hamamoto K: Thallium-201
myocardial tomography with intravenous infusion of adenosine
triphosphate in diagnosis of coronary artery disease. J Am Coll
Cardiol. 26:1196–1201. 1995. View Article : Google Scholar : PubMed/NCBI
|