Huntington's disease (HD) is a lethal autosomal
dominant and progressive neurodegenerative disorder, that is
characterized by motor, cognitive, and behavioral impairment
(1). HD incidence is approximately
5–10 in 100,000 individuals worldwide (2) and encompasses psychiatric symptoms
(e.g., affective disorders, suicide tendency, mania, apathy, and
schizophrenia-like symptoms), cognitive defects (e.g.,
organizational deficit, lack of attention and motor skill learning
deficits), motor impairment (e.g., chorea, rigidity, gait
abnormalities, and bradykinesia), sleep disturbance, and weight
loss (3).
Despite the identification of the gene that is
critical for the pathogenesis of HD as huntingtin (HTT), located in
the short arm of chromosome 4, >20 years ago (4), the development of effective therapies
for HD are is proving to be formidable. Currently, there are no
disease-modifying treatments available other than some approaches
to address certain specific symptoms of HD. Onset of HD symptoms
emerges usually at 35–45 years of age and varies considerably
(5). HD leads to severe brain
atrophy and death, with a clinical course that spans >15–20
years (1). Specifically, striatal
medium spiny neurons (MSNs) of brain appear to be vulnerable in HD,
although potentially other regions of brain can also be affected
(6–8). MSNs are GABAergic neurons, are
predominant in the striatum (9), and
project to the substantia nigra (striatonigral) and globus pallidus
(striatopallidal) (10). It has been
reported that there is a significant loss of approximately 88%
striatal neurons in HD patients as compared to healthy individuals,
even though the precise reasons for this selective vulnerability
and loss of striatal MSNs is not known (11,12).
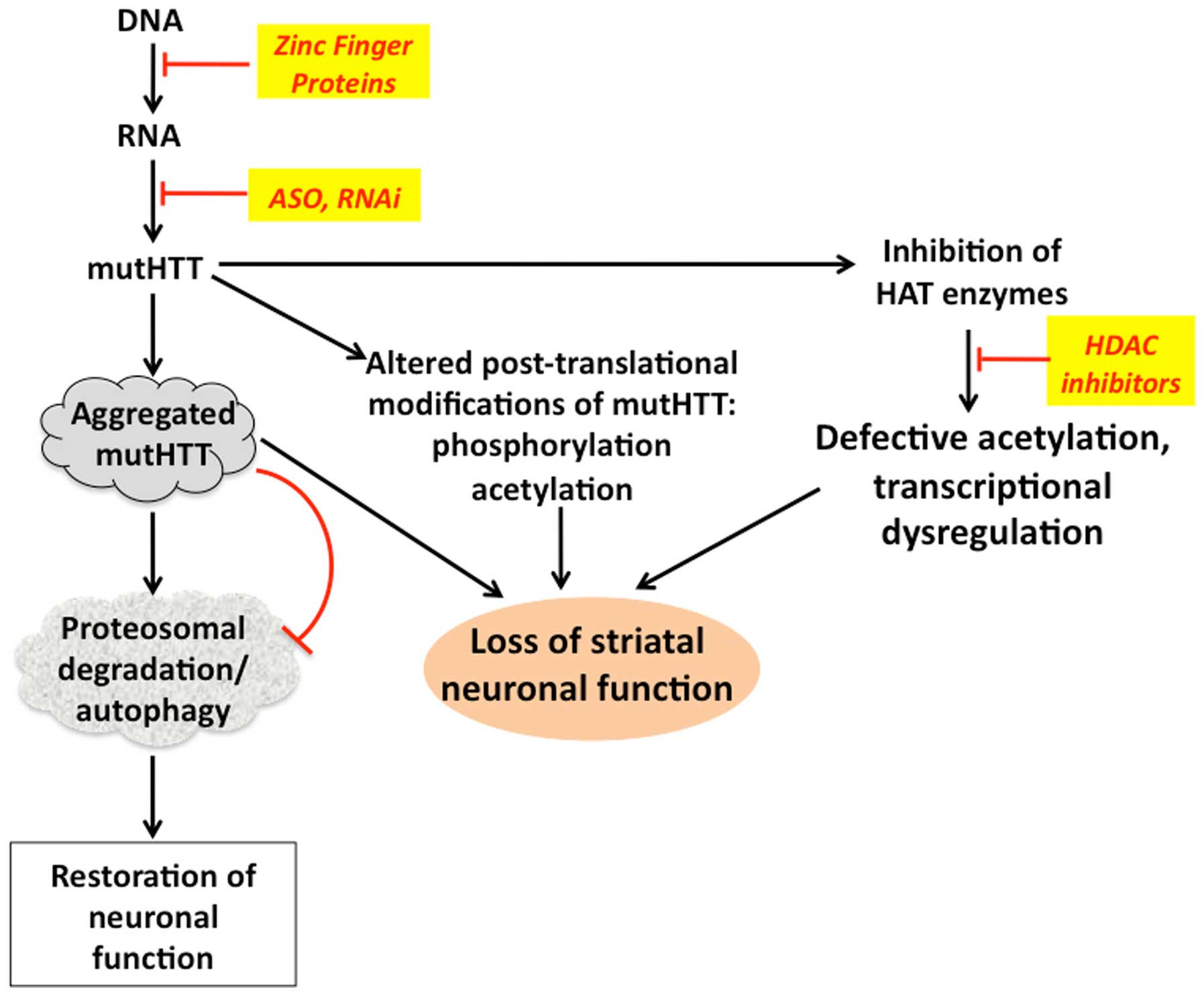
Post-translational modifications of the HTT protein
play an important role in the pathogenesis of HD (Fig. 1). For example, mutHTT is prone to
aggregate in neurons, which is suspected to be part of the
underlying causes of HD. Although mutHTT is ubiquitinylated, its
clearance by the proteosomal system is impaired leading to
accumulation of the aggregates (29). HTT is also likely modified by
phosphorylation, SUMOylation, acetylation and palmitoylation and
these post-translational modifications are important in proper
protein-protein interactions of HTT, which can be significantly
altered by mutations and polyQ additions (30). Histone acetyltransferase (HAT)
enzymes CBP and PCAF were found to be inactivated by mutHTT through
protein-protein interactions, leading to transcriptional and
chromatin remodeling deregulation and contributing to the
pathogenesis of HD (31). It has
been suggested that post-translational modifications be exploited
for therapeutic purposes to enhance the clearance of mutHTT. Thus,
acetylation of the lysine residue K444 in mutHTT enhanced its
clearance via autophagosomes (32),
whereas the phosphorylation of mutHTT at serine 431 and 432 altered
the toxicity and accumulation of mutHTT (33). Phosphorylation of serine residues 13
and 16 reduced its toxicity of mutHTT in vivo (34), whereas phosphorylation at serine 421
restored the ability of mutHTT to promote axonal vesicular
transport and brain-derived neurotrophic factor release (35).
Due to the interaction-mediated inhibitory effects
of mutHTT on HAT enzymes, certain inhibitors of histone
deacetylases (HDAC), in particular HDAC4, have been examined for
their protective effects in some models of HD. The findings showed
that these inhibitors were able to reduce the aggregation of mutHTT
and also rescue the neuronal and corticostriatal synaptic function
(36,37). Notably, acetylation of mutHTT marks
it for ubiquitinylation and subsequent proteosomal degradation and
there is a general decline in chromosomal and protein acetylation
in HD. Thus, inhibition of HDACs, which sustains an elevated level
of protein acetylation, can lead to an increased acetylation status
of mutHTT (38). Inhibitors of other
deacetylase enzymes such as sirtuin 1, and selisistat are shown to
curtail the mutHTT-induced pathology in several model systems
(39) and proved to be safe and
tolerable in recent phase 1B clinical trials (40). Promotion of the proteolytic breakdown
of mHTT through activation of the ubiquitin- proteasome- autophagy
system is another pharmacological approach that is being explored
(41). Thus, promoting autophagy by
inhibiting mTOR with rapamycin, was shown to improve phenotypes in
HD models in Drosophila and mouse (38) and similar effects were observed with
other autophagy-promoting agents (42). Thus, enhancing autophagy to degrade
mutHTT is a viable and important strategy towards HD therapy
(Fig. 2).
Considering that selective modulation of
phosphorylation of serine residues can be exploited to modulate
mutHTT activity, small molecule kinase inhibitors are being tested,
even though their selectivity is being investigated (43). Inasmuch as improper folding and
aggregation of mutHTT is central to the pathogenesis of HD,
attempts are being made to devise cell-permeable chaperones, such
as TCP1-ring complex and ApiCCT1 to selectively prevent the
aggregation of mutHTT and associated toxicity in neuronal cells
(44,45).
Another important HD pathology-associated change is
in the cyclic AMP (cAMP) signaling (46) and aberrant transcription of genes
regulated by the cAMP response element (CRE) (47). Inhibition of phosphodiesterase (PDE)
10A, which regulates cAMP and cyclic guanosine monophosphate
signaling, and is mostly expressed in the MSNs of striatum
(48) is shown to be beneficial
against HD, via restoration of CRE-mediated gene expression
(49). Speciffically, PDE10A
inhibitor-based clinical trials in HD patients are currently
addressing the efficacy and motor functional endpoints (50).
An important signaling pathway that is hyperactive
and contributes to the pathology of HD is MAPK signaling (51). Specifically, overactive c-Jun
N-terminal kinase likely leads to dysregulated axonal transport
(52) and hyperactive p38 may cause
NMDA receptor-mediated excitotoxicity (53). Thus, the overexpression of MKP-1, a
negative modulator of MAPKs, was shown to prevent against
mutHTT-mediated neuronal dysfunction in several models of HD
(54). Similarly, inhibition of
MLK-2 was retarded mutHTT mediated-toxicity (55). NMDA receptor-mediated excitotoxicity,
has been suspected to be an important contributor to HD
pathogenesis and quinolinic acid, an endogenous degradation product
of tryptophan, is a known NMDA receptor agonist. In the pathway of
tryptophan catabolism, kynurenine monooxygenase (KMO) activity
determines the balance between the neuroprotective kynurenic acid
and neurotoxic quinolinic acid. Post-mortem examination of brains
from HD patients revealed that there is an increase in quinolinic
acid and decrease in kynurenic acid. Treatment of HD animal models
with an inhibitor of KMO led to elevated kynurenic acid, as well as
improved survival and striatal neuron function. Recent studies
reported an improved KMO inhibitor, CHDI-340246, which acts only
peripherally and elevates kynurenine and kynurenic acid in HD
rodent and non-human primate models, and protects from neuronal
loss and dysfunction (50,56).
Reducing the content of mutHTT by inhibiting gene
transcription, mRNA translation or promoting the breakdown of mRNA
coding for HTT, may reduce any associated downstream damaging
effects of mutHTT, which otherwise lead to the pathogenesis of HD.
However, considering that loss of HTT protein, even conditionally,
led to neurodegeneration, caution must be exercised to employ
procedures that suppress HTT completely. It is more prudent to
selectively target HTT genes that harbor excessive CAG
repeats, and not normal HTT gene (Figs. 1 and 2).
Inhibition of transcription by zinc finger proteins
(ZFPs) were used to reduce the transcription from the HTT gene.
ZFPs can be designed to allow specific binding to selected DNA
sequences, and are fused to a transcriptional repressor domain, in
order that the gene to which these ZFPs bind, is not expressed, and
thus the corresponding protein production is blocked (57). Using ZFPs it has been observed that
the proximity of the CAG repeat to the 5′ end of the HTT
gene confers selectivity over other genes containing poly-CAG
sequences for targeting with viral vectors for delivering the ZFPs
(58). Such as approach has been
used successfully utilised in mouse model of HD with the resultant
decease in pathological motor manifestations (59,60).
ZFPs have been employed to deliver DNA nucleases to the target
sequences, in a way that excessive CAG repeats are excised from
HTT genes, thus raising the prospect of gene therapy for HD
(61).
Another approach to lower the expression of mutHTT
is to target the corresponding mRNA with specific antisense
oligonucleotides (ASOs), which are single-stranded DNA
oligonucleotides, and bind to complimentary mRNA sequences via
base-pairing and lead to the degradation of the mRNA by RNAse H.
Previous findings have shown that intraventricular infusion of
mutHTT targeting chemically modified ASOs in three separate HD
mouse models was successful in reducing HTT mRNA by 60% and HTT
protein by >80% reduction, in a dose-dependent manner. These
changes were accompanied by delayed mutHTT aggregation and improved
motor performance on a rotarod test. This ASO-induced restoration
of normal functionality was sustained even after the infused ASOs
were removed, indicating that there was a restoration and recovery
of the neurons rendered dysfunctional due to mutHTT (62–64). An
advantage of use of ASO is its broad distribution into different
brain regions following intraventricular infusion. Inasmuch as
mutHTT synthesis is rather ubiquitous, a wider distribution of ASOs
is useful in targeting mutHTT expression and thus curtailing its
deleterious effects (62).
Intrathecal infusion of ASOs for 21 days in non-human primates led
to a sustained decrease by approximately 50% in mutHTT mRNA levels
in frontal cortex, occipital cortex (68%), and spinal cord,
indicating the possible application of ASOs for human situation
(62).
Recent advancements in gene silencing efficiency as
well as sustained long-term effects of RNAi agents have overtaken
the ZFPs and ASOs, despite their therapeutic potential (65). RNAi techniques were successfully
employed to reduce HTT mRNA and protein in in vitro models
of HD (66). Subsequent, in
vivo studies in HD transgenic mice employing AAV-based delivery
of shRNA targeting HTT via single bilateral injections into the
striatum, revealed a significant reduction in mutHTT mRNA and
protein levels, mutHTT aggregates and marked improvement in
behavioral and motor performance parameters (67). This study was followed by several
other in vivo studies using RNAi approach with improvements
in many other HD-associated pathologies (68). Although many of these initial studies
employed pre-symptomatic animal models of HD, subsequent studies
showed that RNAi approaches also reduced the number of mutHTT
inclusions and significantly improved striatal functionality and
motor performance (69). Despite the
success of these gene expression approaches, clinical application
of these methods is not yet feasible and much refinement needs to
be attained in these technologies.
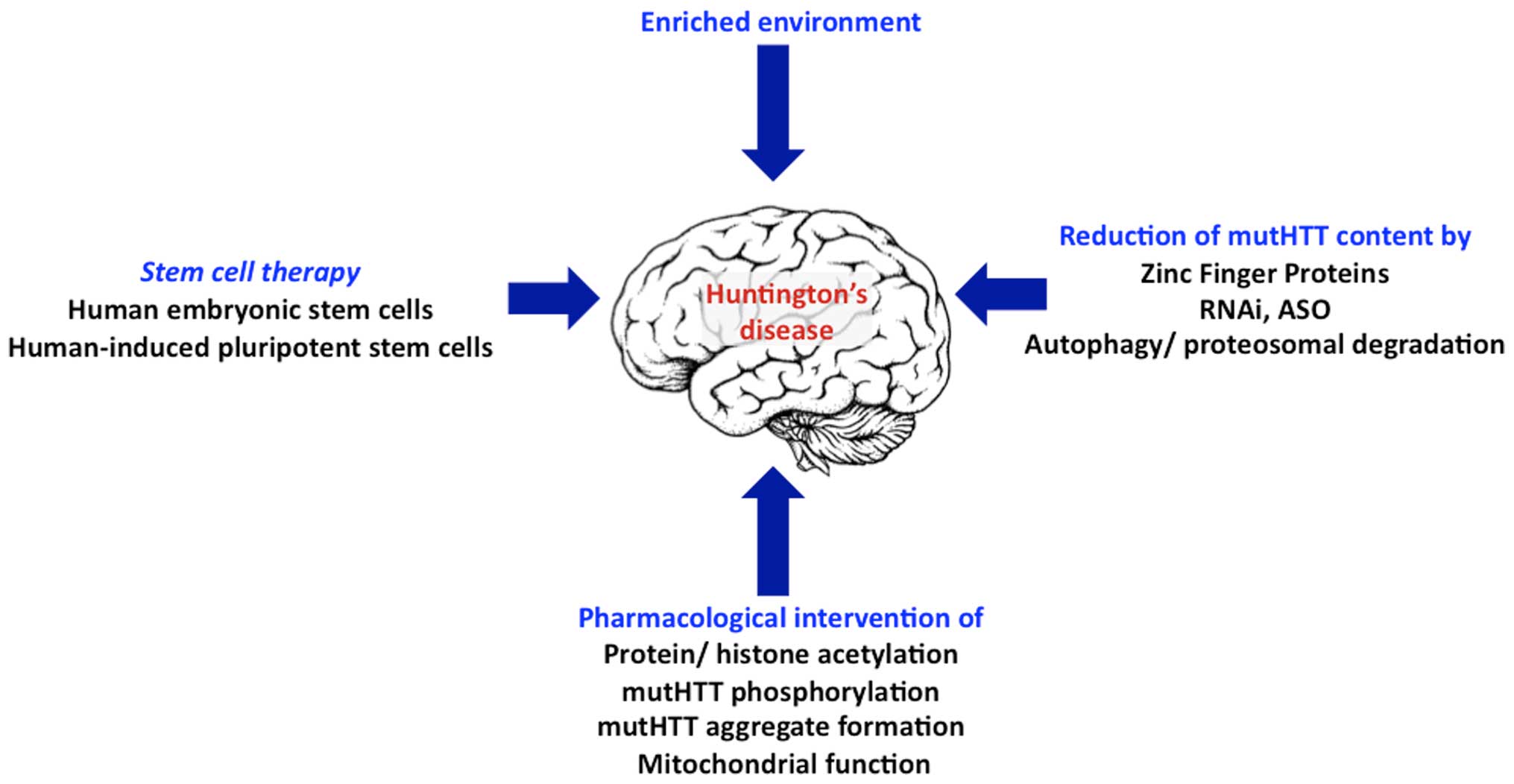
In addition to the abovementioned approaches, stem
cell-based therapies, in particular, using the patient-specific
iPSCs are being developed and are promising to combat HD (Fig. 2) (70).
HD is a hereditary neurodegenerative disorder that
impairs motor and cognitive functions, by targeting striatal MSNs,
with no known cure. MutHTT protein, with an expansion of polyQ
tract is toxic to neurons and is the causative factor of HD.
Therapeutic strategies addressing a reduction in the mutHTT content
at the genome, mRNA or protein degradation level and
post-tranlational modification of mutHTT are being studied in
preclinical models and in clinical trials. Besides the
pharmacological approaches, the use of stem cell therapy, to
replace the lost striatal neurons, is also being examined. These
multiple clinical investigations are promising to identify
therapies that may improve the quality of life for HD patients in
future.
|
1
|
Ross CA, Aylward EH, Wild EJ, Langbehn DR,
Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS,
et al: Huntington disease: Natural history, biomarkers and
prospects for therapeutics. Nat Rev Neurol. 10:204–216. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pringsheim T, Wiltshire K, Day L, Dykeman
J, Steeves T and Jette N: The incidence and prevalence of
Huntington's disease: A systematic review and meta-analysis. Mov
Disord. 27:1083–1091. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bates G, Harper P and Jones L:
Huntington's disease. Oxford University Press; New York, NY:
2002
|
|
4
|
MacDonald M: The Huntington's Disease
Collaborative Research Group: A novel gene containing a
trinucleotide repeat that is expanded and unstable on Huntington's
disease chromosomes. Cell. 72:971–983. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Genetic Modifiers of Huntington's Disease
(GeM-HD) Consortium, . Identification of genetic factors that
modify clinical onset of Huntington's disease. Cell. 162:516–526.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Graveland GA, Williams RS and DiFiglia M:
Evidence for degenerative and regenerative changes in neostriatal
spiny neurons in Huntington's disease. Science. 227:770–773. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mann DM, Oliver R and Snowden JS: The
topographic distribution of brain atrophy in Huntington's disease
and progressive supranuclear palsy. Acta Neuropathol. 85:553–559.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosas HD, Koroshetz WJ, Chen YI, Skeuse C,
Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, et
al: Evidence for more widespread cerebral pathology in early HD: An
MRI-based morphometric analysis. Neurology. 60:1615–1620. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kemp JM and Powell TP: The structure of
the caudate nucleus of the cat: Light and electron microscopy.
Philos Trans R Soc Lond B Biol Sci. 262:383–401. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parent A, Bouchard C and Smith Y: The
striatopallidal and striatonigral projections: Two distinct fiber
systems in primate. Brain Res. 303:385–390. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heinsen H, Strik M, Bauer M, Luther K,
Ulmar G, Gangnus D, Jungkunz G, Eisenmenger W and Götz M: Cortical
and striatal neurone number in Huntington's disease. Acta
Neuropathol. 88:320–333. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vonsattel JP and DiFiglia M: Huntington
disease. J Neuropathol Exp Neurol. 57:369–384. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Myers RH: Huntington's disease genetics.
NeuroRx. 1:255–262. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duyao M, Ambrose C, Myers R, Novelletto A,
Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M,
et al: Trinucleotide repeat length instability and age of onset in
Huntington's disease. Nat Genet. 4:387–392. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trottier Y, Biancalana V and Mandel JL:
Instability of CAG repeats in Huntington's disease: Relation to
parental transmission and age of onset. J Med Genet. 31:377–382.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Telenius H, Kremer B, Goldberg YP,
Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ,
Clarke LA, et al: Somatic and gonadal mosaicism of the Huntington
disease gene CAG repeat in brain and sperm. Nat Genet. 6:409–414.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zielonka D, Piotrowska I and Mielcarek M:
Cardiac dysfunction in huntington's disease. Exp Clin Cardiol.
20:2547–2554. 2014.
|
|
18
|
Zielonka D, Piotrowska I, Marcinkowski JT
and Mielcarek M: Skeletal muscle pathology in Huntington's disease.
Front Physiol. 5:3802014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Imarisio S, Carmichael J, Korolchuk V,
Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood
BR, et al: Huntington's disease: From pathology and genetics to
potential therapies. Biochem J. 412:191–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valor LM: Transcription, epigenetics and
ameliorative strategies in Huntington's Disease: A genome-wide
perspective. Mol Neurobiol. 51:406–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kennedy L, Evans E, Chen CM, Craven L,
Detloff PJ, Ennis M and Shelbourne PF: Dramatic tissue-specific
mutation length increases are an early molecular event in
Huntington disease pathogenesis. Hum Mol Genet. 12:3359–3367. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bečanović K, Nørremølle A, Neal SJ, Kay C,
Collins JA, Arenillas D, Lilja T, Gaudenzi G, Manoharan S, Doty CN,
et al: REGISTRY Investigators of the European Huntington's Disease
Network: A SNP in the HTT promoter alters NF-κB binding and is a
bidirectional genetic modifier of Huntington disease. Nat Neurosci.
18:807–816. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nasir J, Floresco SB, O'Kusky JR, Diewert
VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG and
Hayden MR: Targeted disruption of the Huntington's disease gene
results in embryonic lethality and behavioral and morphological
changes in heterozygotes. Cell. 81:811–823. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeitlin S, Liu JP, Chapman DL, Papaioannou
VE and Efstratiadis A: Increased apoptosis and early embryonic
lethality in mice nullizygous for the Huntington's disease gene
homologue. Nat Genet. 11:155–163. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dragatsis I, Levine MS and Zeitlin S:
Inactivation of Hdh in the brain and testis results in progressive
neurodegeneration and sterility in mice. Nat Genet. 26:300–306.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoffner G, Kahlem P and Djian P:
Perinuclear localization of huntingtin as a consequence of its
binding to microtubules through an interaction with beta-tubulin:
Relevance to Huntington's disease. J Cell Sci. 115:941–948.
2002.PubMed/NCBI
|
|
27
|
Godin JD, Colombo K, MolinaCalavita M,
Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F,
Dragatsis I, et al: Huntingtin is required for mitotic spindle
orientation and mammalian neurogenesis. Neuron. 67:392–406. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
DiFiglia M, SenaEsteves M, Chase K, Sapp
E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, et
al: Therapeutic silencing of mutant huntingtin with siRNA
attenuates striatal and cortical neuropathology and behavioral
deficits. Proc Natl Acad Sci USA. 104:17204–17209. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arrasate M and Finkbeiner S: Protein
aggregates in Huntington's disease. Exp Neurol. 238:1–11. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ehrnhoefer DE, Sutton L and Hayden MR:
Small changes, big impact: Posttranslational modifications and
function of huntingtin in Huntington disease. Neuroscientist.
17:475–492. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zielonka D, Mielcarek M and Landwehrmeyer
GB: Update on Huntington's disease: Advances in care and emerging
therapeutic options. Parkinsonism Relat Disord. 21:169–178. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jeong H, Then F, Melia TJ Jr, Mazzulli JR,
Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, et al:
Acetylation targets mutant huntingtin to autophagosomes for
degradation. Cell. 137:60–72. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dong G, Callegari E, Gloeckner CJ, Ueffing
M and Wang H: Mass spectrometric identification of novel
posttranslational modification sites in Huntingtin. Proteomics.
12:2060–2064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gu X, Greiner ER, Mishra R, Kodali R,
Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R and Yang
XW: Serines 13 and 16 are critical determinants of full-length
human mutant huntingtin induced disease pathogenesis in HD mice.
Neuron. 64:828–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zala D, Colin E, Rangone H, Liot G,
Humbert S and Saudou F: Phosphorylation of mutant huntingtin at
S421 restores anterograde and retrograde transport in neurons. Hum
Mol Genet. 17:3837–3846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Venuto CS, McGarry A, Ma Q and Kieburtz K:
Pharmacologic approaches to the treatment of Huntington's disease.
Mov Disord. 27:31–41. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mielcarek M, Benn CL, Franklin SA, Smith
DL, Woodman B, Marks PA and Bates GP: SAHA decreases HDAC 2 and 4
levels in vivo and improves molecular phenotypes in the R6/2 mouse
model of Huntington's disease. PLoS One. 6:e277462011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ravikumar B, Vacher C, Berger Z, Davies
JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et
al: Inhibition of mTOR induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse models of Huntington
disease. Nat Genet. 36:585–595. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Smith MR, Syed A, Lukacsovich T, Purcell
J, Barbaro BA, Worthge SA, Wei SR, Pollio G, Magnoni L, Scali C, et
al: A potent and selective Sirtuin 1 inhibitor alleviates pathology
in multiple animal and cell models of Huntington's disease. Hum Mol
Genet. 23:2995–3007. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Reilmann R, Squitieri F, Priller J, Saft
C, Mariotti C, Suessmuth S, Nemeth A, Tabrizi S, Quarrell O,
Craufurd D, et al: Safety and tolerability of selisistat for the
treatment of huntington's disease: Results from a randomized,
double-blind, placebo-controlled phase II trial. Neurology. (Suppl
10)82:S47.0042014.
|
|
41
|
Labbadia J and Morimoto RI: Huntington's
disease: Underlying molecular mechanisms and emerging concepts.
Trends Biochem Sci. 38:378–385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Renna M, JimenezSanchez M, Sarkar S and
Rubinsztein DC: Chemical inducers of autophagy that enhance the
clearance of mutant proteins in neurodegenerative diseases. J Biol
Chem. 285:11061–11067. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Atwal RS, Desmond CR, Caron N, Maiuri T,
Xia J, Sipione S and Truant R: Kinase inhibitors modulate
huntingtin cell localization and toxicity. Nat Chem Biol.
7:453–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tam S, Geller R, Spiess C and Frydman J:
The chaperonin TRiC controls polyglutamine aggregation and toxicity
through subunit-specific interactions. Nat Cell Biol. 8:1155–1162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sontag EM, Joachimiak LA, Tan Z, Tomlinson
A, Housman DE, Glabe CG, Potkin SG, Frydman J and Thompson LM:
Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates
mutant Huntingtin cellular phenotypes. Proc Natl Acad Sci USA.
110:3077–3082. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gines S, Seong IS, Fossale E, Ivanova E,
Trettel F, Gusella JF, Wheeler VC, Persichetti F and MacDonald ME:
Specific progressive cAMP reduction implicates energy deficit in
presymptomatic Huntington's disease knock-in mice. Hum Mol Genet.
12:497–508. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sugars KL, Brown R, Cook LJ, Swartz J and
Rubinsztein DC: Decreased cAMP response element-mediated
transcription: An early event in exon 1 and full-length cell models
of Huntington's disease that contributes to polyglutamine
pathogenesis. J Biol Chem. 279:4988–4999. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Coskran TM, Morton D, Menniti FS,
Adamowicz WO, Kleiman RJ, Ryan AM, Strick CA, Schmidt CJ and
Stephenson DT: Immunohistochemical localization of
phosphodiesterase 10A in multiple mammalian species. J Histochem
Cytochem. 54:1205–1213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kleiman RJ, Kimmel LH, Bove SE, Lanz TA,
Harms JF, Romegialli A, Miller KS, Willis A, des Etages S, Kuhn M,
et al: Chronic suppression of phosphodiesterase 10A alters striatal
expression of genes responsible for neurotransmitter synthesis,
neurotransmission, and signaling pathways implicated in
Huntington's disease. J Pharmacol Exp Ther. 336:64–76. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wild EJ and Tabrizi SJ: Targets for future
clinical trials in Huntington's disease: What's in the pipeline?
Mov Disord. 29:1434–1445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gianfriddo M, Melani A, Turchi D,
Giovannini MG and Pedata F: Adenosine and glutamate extracellular
concentrations and mitogen-activated protein kinases in the
striatum of Huntington transgenic mice. Selective antagonism of
adenosine A2A receptors reduces transmitter outflow. Neurobiol Dis.
17:77–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Morfini GA, You YM, Pollema SL, Kaminska
A, Liu K, Yoshioka K, Björkblom B, Coffey ET, Bagnato C, Han D, et
al: Pathogenic huntingtin inhibits fast axonal transport by
activating JNK3 and phosphorylating kinesin. Nat Neurosci.
12:864–871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fan J, Gladding CM, Wang L, Zhang LY,
Kaufman AM, Milnerwood AJ and Raymond LA: P38 MAPK is involved in
enhanced NMDA receptor-dependent excitotoxicity in YAC transgenic
mouse model of Huntington disease. Neurobiol Dis. 45:999–1009.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Taylor DM, Moser R, Régulier E, Breuillaud
L, Dixon M, Beesen AA, Elliston L, Silva Santos MF, Kim J, Jones L,
et al: MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in
Huntington's disease via additive effects of JNK and p38
inhibition. J Neurosci. 33:2313–2325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Apostol BL, Simmons DA, Zuccato C, Illes
K, Pallos J, Casale M, Conforti P, Ramos C, Roarke M, Kathuria S,
et al: CEP-1347 reduces mutant huntingtin-associated neurotoxicity
and restores BDNF levels in R6/2 mice. Mol Cell Neurosci. 39:8–20.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mrzljak L: A05 targeting kmo: Basic
understanding and gaps. J Neurol Neurosurg Psychiatry. 85:A22014.
View Article : Google Scholar
|
|
57
|
Papworth M, Kolasinska P and Minczuk M:
Designer zinc-finger proteins and their applications. Gene.
366:27–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jiang H, Sun YM, Hao Y, Yan YP, Chen K,
Xin SH, Tang YP, Li XH, Jun T, Chen YY, Liu ZJ, Wang CR, Li H, Pei
Z, Shang HF, Zhang BR, Gu WH, Wu ZY, Tang BS and Burgunder JM:
Huntingtin gene CAG repeat numbers in Chinese patients with
Huntington's disease and controls. Eur J Neurol. 21:637–642. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
GarrigaCanut M, Agustín-Pavón C, Herrmann
F, Sánchez A, Dierssen M, Fillat C and Isalan M: Synthetic zinc
finger repressors reduce mutant huntingtin expression in the brain
of R6/2 mice. Proc Natl Acad Sci USA. 109:E3136–E3145. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zeitler J, Pearl JR, Froelich S, Yu Q,
Paschon DE, Miller JC, Marlen K, Guschin D, Narayanan A, Zhang L,
et al: Allele- specific repression of mutant Huntingtin expression
by engineered zinc finger transcriptional repressors as a potential
therapy for Huntington's disease. PNAS. 108:7052–7057.
2011.PubMed/NCBI
|
|
61
|
Li H, Haurigot V, Doyon Y, Li T, Wong SY,
Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L, et al: In
vivo genome editing restores haemostasis in a mouse model of
haemophilia. Nature. 475:217–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kordasiewicz HB, Stanek LM, Wancewicz EV,
Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH,
Shihabuddin LS, et al: Sustained therapeutic reversal of
Huntington's disease by transient repression of huntingtin
synthesis. Neuron. 74:1031–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lu XH and Yang XW: ‘Huntingtin holiday’:
Progress toward an antisense therapy for Huntington's disease.
Neuron. 74:964–966. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Carroll JB, Warby SC, Southwell AL, Doty
CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM and Hayden
MR: Potent and selective antisense oligonucleotides targeting
single-nucleotide polymorphisms in the Huntington disease
gene/allele-specific silencing of mutant huntingtin. Mol Ther.
19:2178–2185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Miyagishi M, Hayashi M and Taira K:
Comparison of the suppressive effects of antisense oligonucleotides
and siRNAs directed against the same targets in mammalian cells.
Antisense Nucleic Acid Drug Dev. 13:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen ZJ, Kren BT, Wong PY, Low WC and
Steer CJ: Sleeping Beauty-mediated down-regulation of huntingtin
expression by RNA interference. Biochem Biophys Res Commun.
329:646–652. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Harper SQ, Staber PD, He X, Eliason SL,
Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL and Davidson BL:
RNA interference improves motor and neuropathological abnormalities
in a Huntington's disease mouse model. Proc Natl Acad Sci USA.
102:5820–5825. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Godinho BM, Malhotra M, O'Driscoll CM and
Cryan JF: Delivering a disease-modifying treatment for Huntington's
disease. Drug Discov Today. 20:50–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Drouet V, Perrin V, Hassig R, Dufour N,
Auregan G, Alves S, Bonvento G, Brouillet E, LuthiCarter R,
Hantraye P, et al: Sustained effects of nonallele-specific
Huntingtin silencing. Ann Neurol. 65:276–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Golas MM and Sander B: Use of human stem
cells in Huntington disease modeling and translational research.
Exp Neurol. 278:76–90. 2016. View Article : Google Scholar : PubMed/NCBI
|