Introduction
Andersen-Tawil syndrome (ATS), also known as
Andersen syndrome and long QT syndrome 7, is a rare channelopathy
inherited in an autosomal dominant fashion (1,2). ATS is
characterized by the triad of episodic flaccid muscle weakness
(periodic paralysis may be hypo-, hyper- or normokalemic); cardiac
arrhythmias and dysmorphic features, including low-set ears,
hypertelorism, small mandible, clinodactyly, syndactyly, short
stature and scoliosis (3,4). In addition, hypoplastic kidney and
valvular heart disease have been reported in patients with ATS
(5). There are two types of ATS.
Type 1 ATS accounts for ~60% of patients with the disorder and is
caused by mutations in the potassium channel inwardly rectifying
subfamily J member 2 (KCNJ2) gene, which alters the normal
structure and function of potassium channels, or prevents the
channels from being inserted correctly into the cell membrane
(6). This disrupts the flow of
potassium ions in skeletal and cardiac muscle, leading to the
periodic paralysis and irregular heart rhythm characteristics
(6). The remaining 40% of cases are
designated as type 2 ATS, the cause of which remains unknown
(7).
Myasthenia gravis (MG) is an autoimmune disorder of
the neuromuscular junction. The hallmark of MG is fatigability.
Muscles become progressively weaker during periods of activity and
improve after rest. Muscles that control eye and eyelid movement,
facial expressions, chewing, talking and swallowing are
particularly susceptible (8,9). In addition, the muscles that control
breathing and neck and limb movements can be affected. Symptoms may
include asymmetrical ptosis, diplopia, unstable or waddling gait,
weakness in the arms, hands, fingers, legs, and neck, a change in
facial expression, dysphagia, shortness of breath and dysarthria
(8). A myasthenic crisis may require
assisted ventilation to sustain life when paralysis of the
respiratory muscles occurs. Myasthenia does not directly affect
cardiac muscle (10).
Anti-acetylcholine receptor antibodies are considered to be
pathognomonic and pathogenetic for MG, as they block acetylcholine
receptors at the postsynaptic neuromuscular junction (9), inhibiting the excitatory effects of the
neurotransmitter acetylcholine on nicotinic receptors at
neuromuscular junctions.
The present study describes a case of concomitant
presentation of ATS and MG. To the best of our knowledge, such a
case has not been reported previously in the literature and
represents a diagnostic and management challenge.
Case report
A 31-year-old woman with a history of morbid obesity
(body mass index, 58; weight, 375 lbs; height, 5′7′′) and periodic
weakness was admitted to a tertiary University Hospital and
intubated in August 2008 due to hemodynamic instability and
cardiogenic shock. The patient exhibited facial anomalies,
including hypertelorism, ptosis, widely spaced eyes, low-set ears
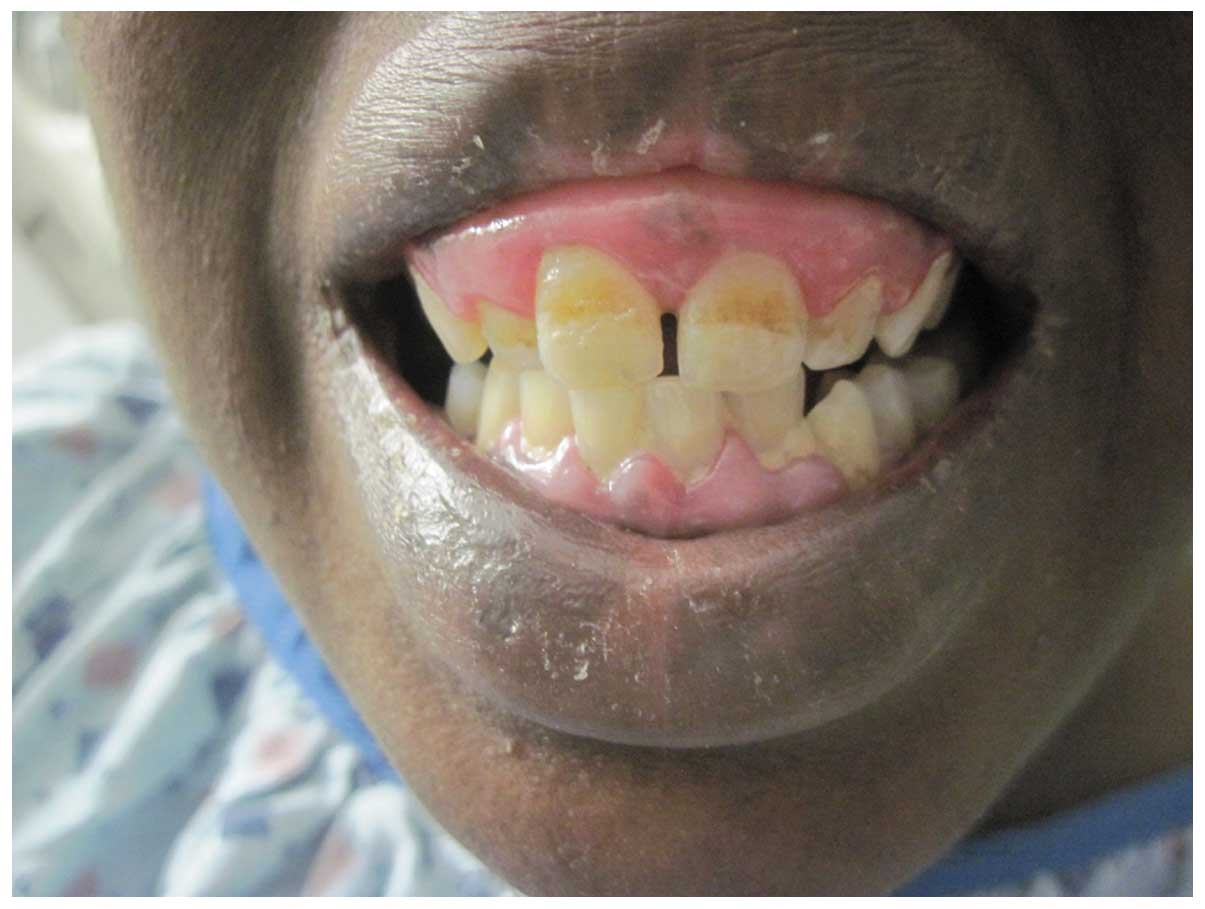
and micrognathia, and dental abnormalities (Fig. 1). Laboratory results at admission
demonstrated hypokalemia (K+, 2.5 mEq/l; normal range, 3.5–5.0
mEq/l) and acute renal insufficiency (creatinine, 1.7 mg; normal in
females, 0.6–1.1 mg), but were otherwise unremarkable.
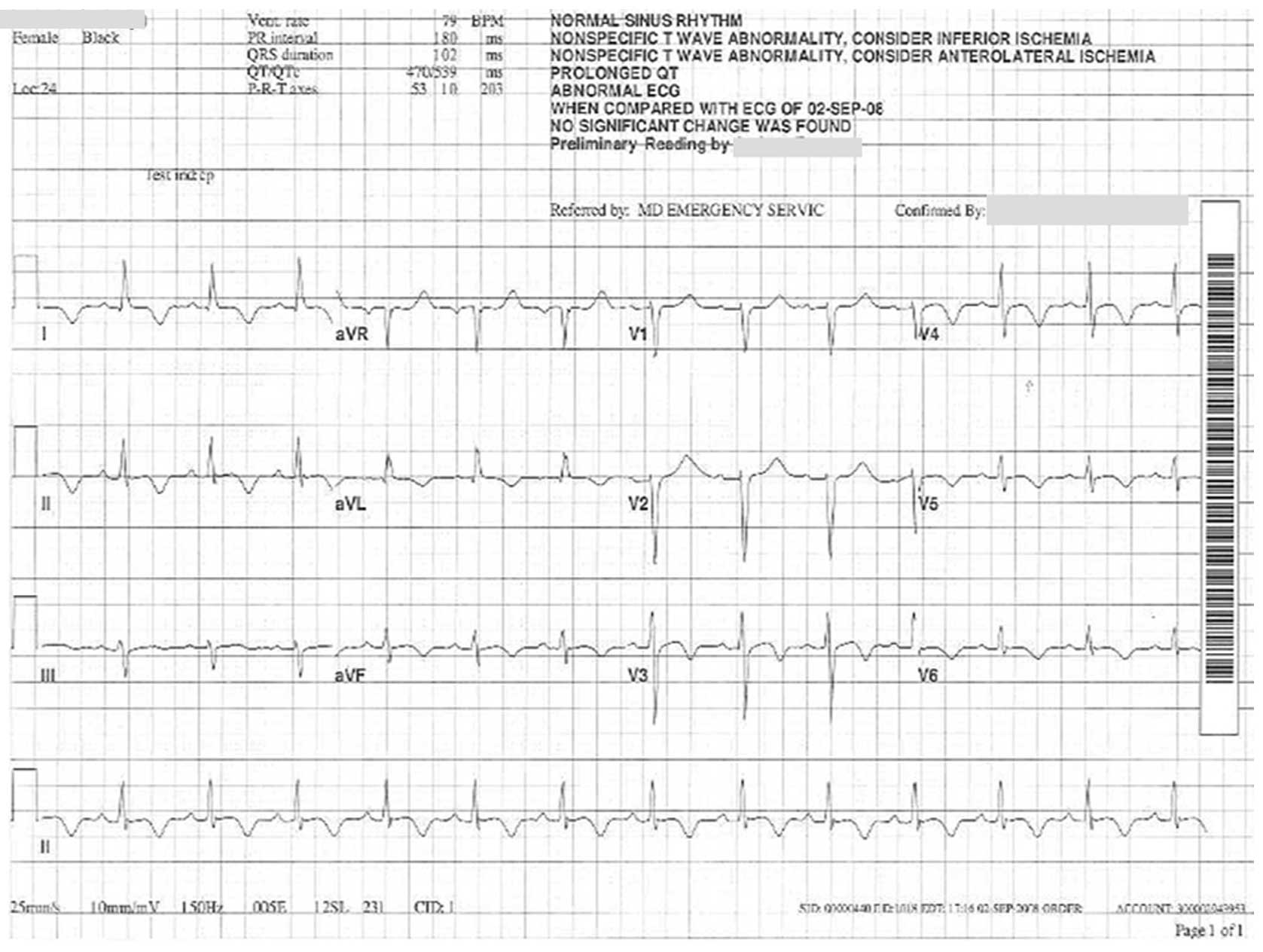
Electrocardiography indicated a prolonged QT-interval (QTc, 539 ms;
normal, <470 ms for females), ST-elevation in the inferior and
anterolateral leads (1 mm), and subsequent third-degree
atrioventricular block (Fig. 2).
Clinical findings were consistent with those of ATS. No mutations
of the KCNJ2 gene were detected by sequencing analysis, as
previously described in this case (3). Although the patient was medically
stabilized with an implanted pacemaker and normalized electrolytes,
her weakness did not improve.
Neurological examination revealed that the patient
was mentally competent but experienced generalized weakness with
bilateral ptosis, binocular diplopia in all directions, dysarthria
with a lax jaw and poor dentition. The patient was unable to close
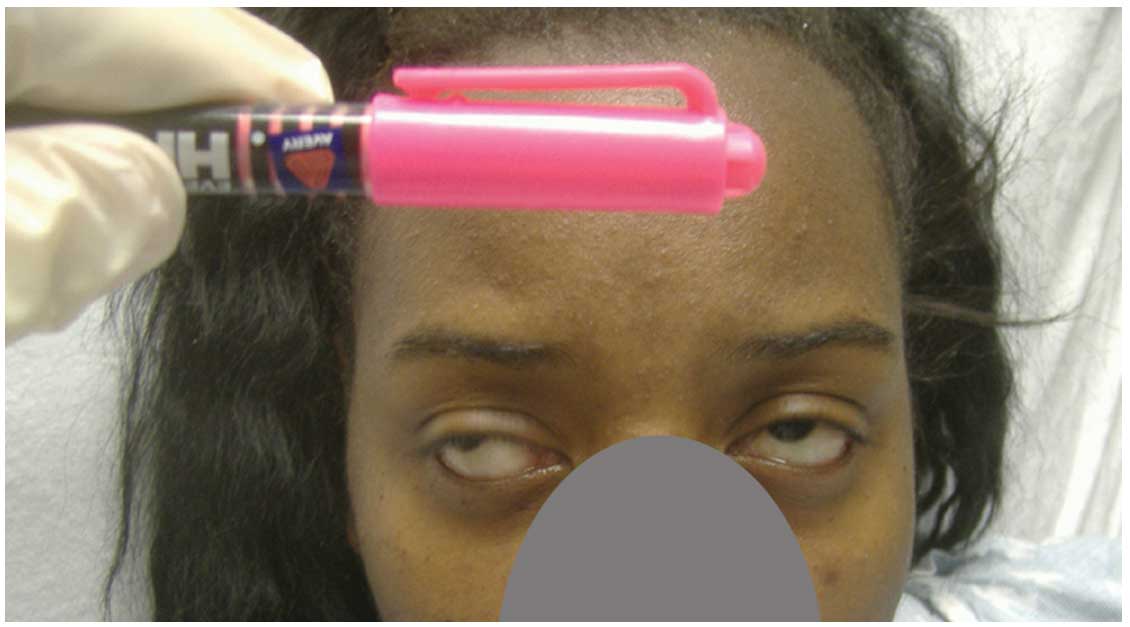
her eyes completely or hold up her head. The fatigue test by upper
gazing, as described previously (11), provoked dysconjugated eyes (Fig. 3). The patient's muscle strength was
weak in the lower (2/5) and upper (3/5) extremities, as determined
by the criteria outlined by the Medical Research Council
(https://www.mrc.ac.uk/documents/pdf/complex-interventions-guidance/).
Tendon reflexes and sensory responses (including pinprick,
temperature and vibration) were normal. No muscle tenderness or
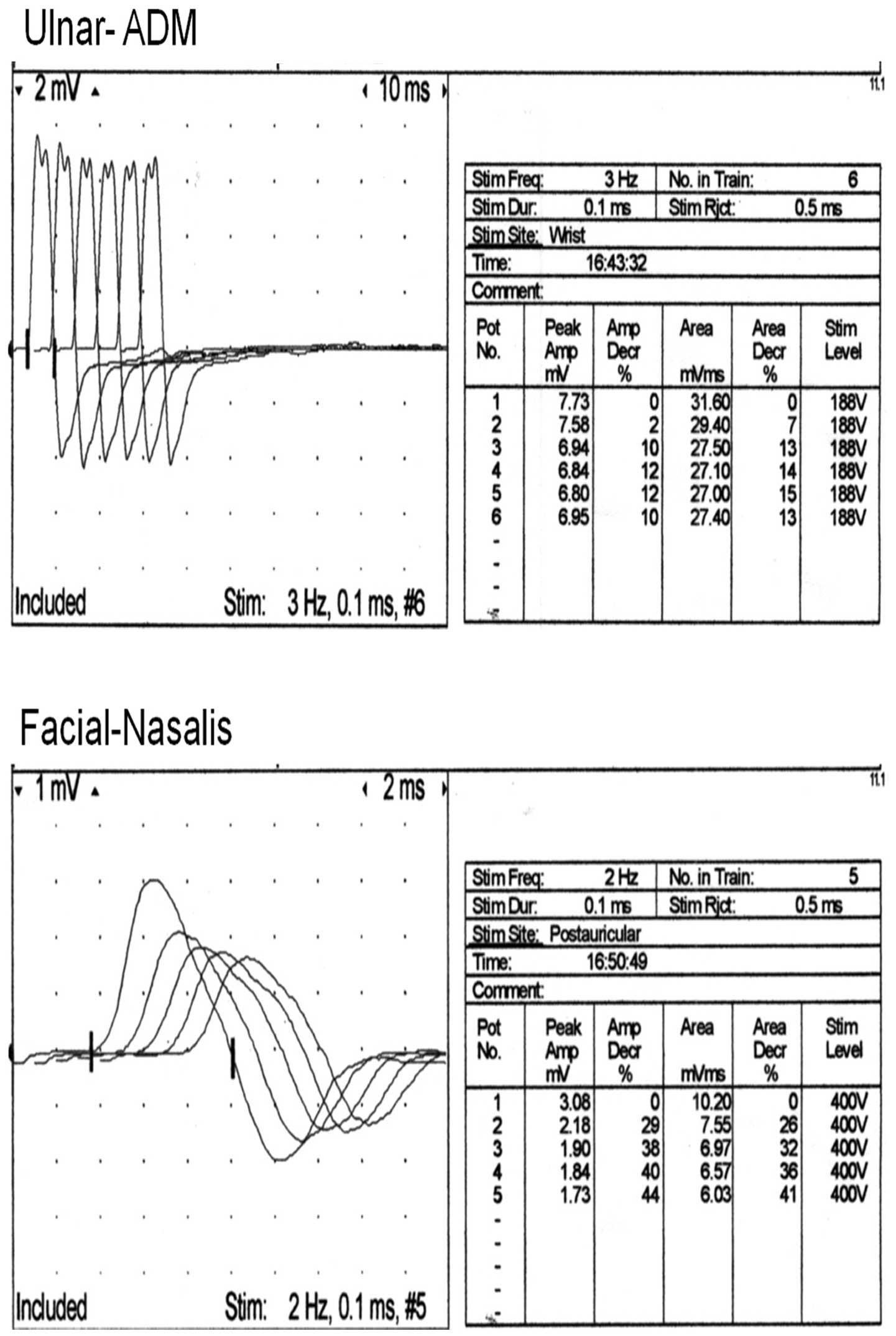
pathologic reflexes were detected. Repetitive nerve stimulation of
the ulnar-abductor digiti minimi muscle and nasalis facial muscle
showed abnormal decremental responses (Fig. 4). The expression level of anti-AChR
antibody (performed by Quest Diagnostics, Philadelphia, PA, USA)
was significantly elevated (178 nmol/l; normal, <0.3 nmol/l).
The patient was treated with 0.4 g/kg/day intravenous
immunoglobulin (IVIG; GAMMAGARD LIQUID; Baxalter US Inc.,
Bannockburn, IL, USA) for 5 days, as well as 60 mg pyridostigmine
three times per day, and her symptoms improved.
In the following year, it transpired at follow-up
that the patient had experienced episodic events of weakness which
were initially considered as myasthenic crises, and the patient was
admitted into the Intensive Care Unit and provided further IVIG
treatment (0.4 g/kg/day for 5 days). The cause of these events was
eventually identified as being associated with potassium levels
<3.2 mEq/l. The patient was subsequently treated with oral
potassium supplementation (20 mEq/day) and pyridostigmine (90 mg
four times per day), and her symptoms improved. Follow-up at 6
years demonstrated that the patient remained in a stable condition
without any further weakness events. Written informed consent was
obtained for the present study.
Discussion
In the current case report, a rare case of a 31-year
old woman with concurrent ATS and MG is presented. To the best of
our knowledge, this has not been previously reported in the
literature. ATS is a rare genetic disorder of unknown prevalence
which is associated with <10% primary periodic paralysis cases.
The precise incidence of periodic paralysis remains unknown,
however it is estimated to be ~1:100,000. At least 50% of
individuals diagnosed with ATS have an affected parent (12).
ATS should be considered in any individual
presenting with periodic paralysis and ventricular arrhythmias.
Individuals with episodic weakness or cardiac symptoms require
careful evaluation by a neurologist and/or cardiologist,
measurement of the serum potassium concentration (including
baseline measurements and measurements during attacks of flaccid
paralysis), a 12-lead electrocardiogram and 24-h Holter monitoring.
Differential diagnosis depends on the initial presentation and
includes primary and secondary periodic paralyses and thyrotoxic
periodic paralysis (3,4). The results of routine nerve conduction
analyses are typically normal between episodes in patients with
ATS. A more sensitive neuroelectrophysiological study may reveal an
immediate post-exercise increment followed by an abnormal decrement
in the compound motor action potential amplitude (>40%)
(13) or area (>50%) at 20 to 40
min post-exercise (14,15). In a previous study of 11 individuals
with ATS, 82% met long-exercise amplitude decrement criteria for
abnormal testing (16).
The presence of a pathogenic KCNJ2 sequence variant
confirms the diagnosis of ATS1 (5,7).
Sequence analysis and mutation scanning of the entire gene can
result in similar mutation detection frequencies; however, mutation
detection rates for mutation scanning may vary considerably among
laboratories, depending on the specific protocol used. KCNJ2 is the
only gene in which mutations are known to cause ATS1. The mutation
p.Arg218Trp is considered a potential hotspot for disease-causing
mutations (4,17,18).
However, ~40% of ATS cases are without KCNJ2 gene defects, and
their diagnoses may be more difficult (4,17).
MG occurs in all ethnic groups and both sexes.
Although MG most commonly affects women aged <40 years followed
by individuals aged between 50 and 70 years old of either sex, it
has been known to occur at any age (8). Diagnosis of MG may be delayed if the
symptoms are subtle or variable, rendering it hard to distinguish
between normal variants and other neurological disorders (8). A detailed history and neurological
examination may reveal easy fatigability, with the weakness
improving with rest and worsening again during subsequent exertion
testing. A good response to medication, such as an
acetylcholinesterase inhibitor (pyridostigmine), may also indicate
MG.
The present study described the case of a
31-year-old woman who presented with episodic weakness, cardiac
arrhythmias and dysmorphic features. Clinical findings were
consistent with those of type 2 ATS, in which the KCNJ2 gene
mutation is absent. Notably, the patient exhibited decreased muscle
strength with abnormal decremental responses on repetitive nerve
stimulation, which indicated neuromuscular transmissional
dysfunction. The levels of anti-AChR antibody were tested and
significant elevation was detected, which confirmed the diagnosis
of MG in addition to ATS in the patient. Diagnosis of concurrent
ATS and MG may be overlooked due to the prevalence of muscle
weakness. The patient's cardiac condition was well controlled
following the implantation of a cardiac pacer. Although the
patient's weakness initially responded well to an
antiacetylcholinesterase agent (pyridostigmine), significant
episodic weakness was triggered by hypokalemia, which may result in
a clinical management challenge if the hypokalemia is not
recognized. Notably, episodic flaccid muscle weakness in ATS with
periodic paralysis may be hypo-, hyper- or normokalemic. The
patient described in the current case study has remained in a
stable condition with a normal quality of life and no recurrence of
episodic weakness in 6 years, whilst being treated with a regimen
of oral potassium supplementation and pyridostigmine.
In conclusion, although it is extremely rare,
concomitant presentation of ATS and MG, which have very different
pathogeneses, may occur in a single individual. Managing a patient
with concurrent ATS and MG may become a diagnostic and therapeutic
challenge. Therefore, we recommend that an investigation of MG
should be performed in patients with ATS.
References
|
1
|
Andersen ED, Krasilnikoff PA and Overvad
H: Intermittent muscular weakness, extrasystoles and multiple
developmental anomalies, A new syndrome? Acta paediatr Scand.
60:559–564. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tawil R, Ptacek LJ, Pavlakis SG, DeVivo
DC, Penn AS, Ozdemir C and Griggs RC: Andersen's syndrome:
Potassium-sensitive periodic paralysis, ventricular ectopy, and
dysmorphic features. Ann Neurol. 35:326–330. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang G, Knight L, Ji R, Lawrence P, Kanaan
U, Li L, Das A, Cui B, Zou W, Penny DJ and Fan Y: Early onset
severe pulmonary arterial hypertension with ‘two-hit’
digenicmutations in both BMPR2 and KCNA5 genes. Int J Cardiol.
177:e167–e169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hosaka Y, Hanawa H, Washizuka T, Chinushi
M, Yamashita F, Yoshida T, Komura S, Watanabe H and Aizawa Y:
Function, subcellular localization and assembly of a novel mutation
of KCNJ2 in Andersen's syndrome. J Mol Cell Cardiol. 35:409–415.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tristani-Firouzi M, Jensen JL, Donaldson
MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H,
Fidzianska A, Plaster N, et al: Functional and clinical
characterization of KCNJ2 mutations associated with LQT7 (Andersen
syndrome). J Clin Invest. 110:381–388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pegan S, Arrabit C, Slesinger PA and Choe
S: Andersen's syndrome mutation effects on the structure and
assembly of the cytoplasmic domains of Kir2.1. Biochemistry.
45:8599–8606. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim JB and Chung KW: Novel de novo
mutation in the KCNJ2 gene in a patient with andersen-tawil
syndrome. Pediatric Neurology. 41:464–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scherer K, Bedlack RS and Simel DL: Does
this patient have myasthenia gravis? JAMA. 293:1906–1914. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Conti-Fine BM, Milani M and Kaminski HJ:
Myasthenia gravis: Past, present, and future. J Clin Invest.
116:2843–2854. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bedlack RS and Sanders DB: How to handle
myasthenic crisis Essential steps in patient care. Postgrad Med.
107:220–222. 2000. View Article : Google Scholar
|
|
11
|
Kittiwatanapaisan W, Gauthier DK, Williams
AM and Oh SJ: Fatigue in Myasthenia Gravis patients. J Neurosci
Nurs. 35:87–93. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Statland JM, Tawil R and Venance SL:
Andersen-Tawil Syndrome. GeneReviews® (Internet),
Seattle (WA) University of Washington, Seattle. 2004.http://www.ncbi.nlm.nih.gov/books/NBK1264/June
23–2015
|
|
13
|
Katz JS, Wolfe GI, Iannaccone S, Bryan WW
and Barohn RJ: The exercise test in Andersen syndrome. Arch Neurol.
56:352–356. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuntzer T, Flocard F, Vial C, Kohler A,
Magistris M, Labarre-Vila A, Gonnaud PM, Ochsner F, Soichot P, Chan
V and Monnier G: Exercise test in muscle channelopathies and other
muscle disorders. Muscle Nerve. 23:1089–1094. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fournier E, Arzel M, Sternberg D, Vicart
S, Laforet P, Eymard B, Willer JC, Tabti N and Fontaine B:
Electromyography guides toward subgroups of mutations in muscle
channelopathies. Ann Neurol. 56:650–661. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan SV, Matthews E, Barber M, Burge JA,
Rajakulendran S, Fialho D, Sud R, Haworth A, Koltzenburg M and
Hanna MG: Refined exercise testing can aid DNA-based diagnosis in
muscle channelopathies. Ann Neurol. 69:328–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Plaster NM, Tawil R, Tristani-Firouzi M,
Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt
E, Barohn R, et al: Mutations in Kir2.1 cause the developmental and
episodic electrical phenotypes of Andersen's syndrome. Cell.
105:511–519. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davies NP, Imbrici P, Fialho D, Herd C,
Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman
BR, et al: Andersen-Tawil syndrome: New potassium channel mutations
and possible phenotypic variation. Neurology. 65:1083–1094. 2005.
View Article : Google Scholar : PubMed/NCBI
|