Introduction
Epidermolysis bullosa simplex (EBS) refers to a
number of inherited disorders characterized by mechanical
stress-induced blistering of the skin (1). EBS comprises three primary types:
Localized [EBS-loc; Online Mendelian Inheritance in Man (OMIM) no.
131800], generalized severe (EBS-gen sev; OMIM no. 131760) and
generalized intermediate (EBS-gen intermed; OMIM no. 131900)
(1). The ultrastructural
pathogenesis of EBS-gen sev is the collapse of keratin filaments in
basal epidermal cells, resulting in basal cell cytolysis and
sequent intraepidermal blister formation, which can differ from
other subtypes (2–4). EBS-gen sev typically presents with
characteristic features of large, generalized blisters in early
infants, and small, clustered (herpetiform) blisters in childhood.
In neonates and infants, EBS-gen sev is life-threatening as
cutaneous lesions are typically severe, resulting in difficulties
in feeding and care (5–7). Subsequent to infancy, particularly
during late childhood and adulthood, the prognosis is favorable
(5–7). Mutations located at the highly
conserved α-helical end segments of the 1A domain of keratin 5
(KRT5) and 2B domain of KRT14, also known as helix
initiation peptide (HIP) and helix termination peptide (HTP),
respectively, typically result in EBS-gen sev (8,9). To
date, four pathogenic mutations have been reported to be
responsible for EBS-gen sev in the Chinese population, including
p.Arg165Ser and p.Lys199Asn in KRT5, and p.Arg125Cys and
p.Arg125His in KRT14 (10–13).
In the present study, molecular genetic tests were
performed in a 2-year-old boy with suspected EBS, and a novel
missense mutation c.503A>G (p.Glu168Gly) located at the
N-terminal end segment of the 1A domain of KRT5 (HIP)
confirmed a diagnosis of EBS-gen sev.
Materials and methods
Case
The proband was a 2-year-old boy that was referred
to the Department of Dermatology of Xinhua Hospital (Shanghai,
China) in October 2013. The patient had been reported to present
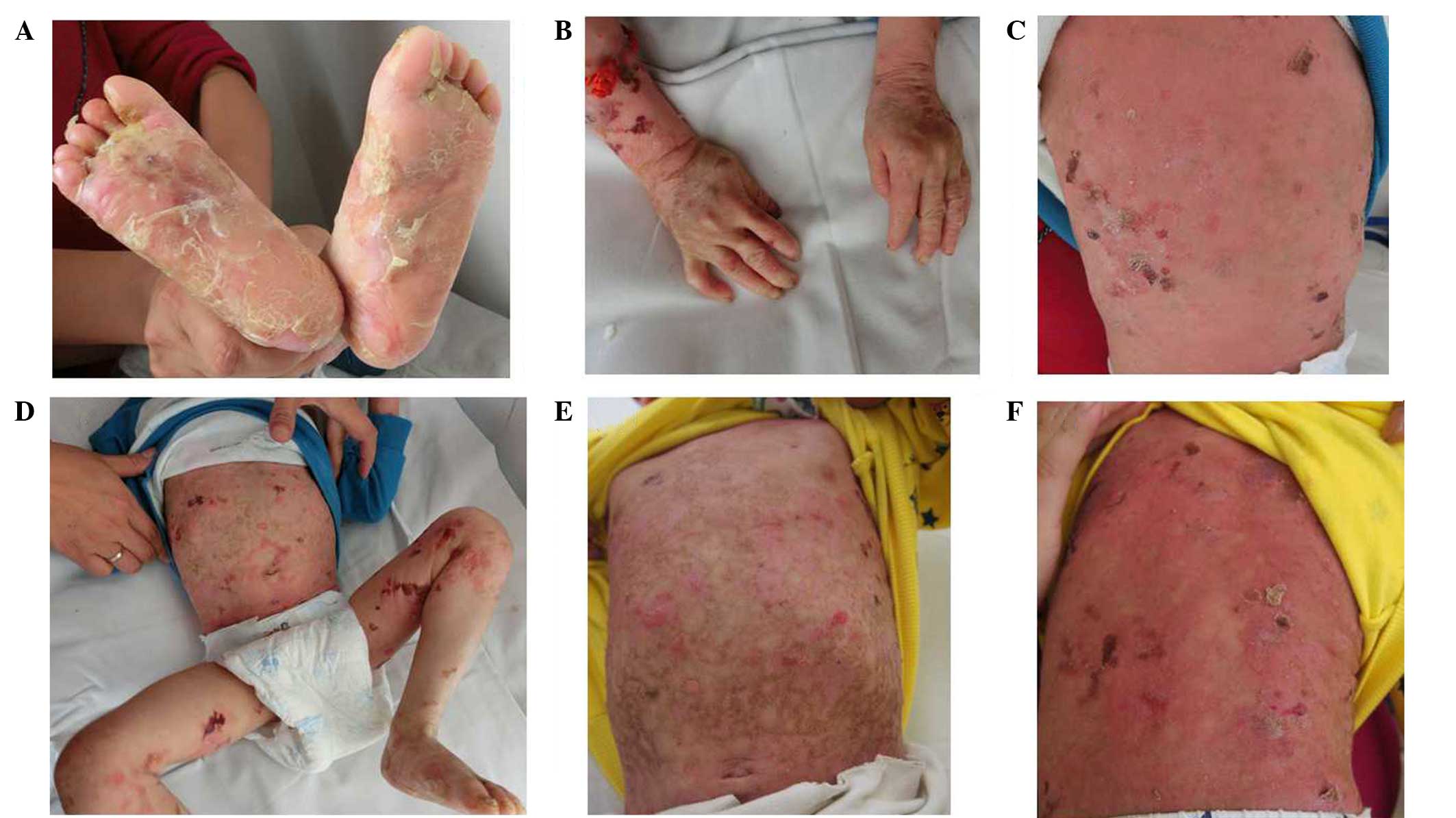
generalized blisters throughout his body since birth (Fig. 1A-D), which were worsened by friction
or trauma and were not treated. No other symptoms were observed
other than blisters. At the 9-month follow-up visit a
dermatological examination showed that there was no evident
improvement (Fig. 1E and F). No
other relevant medical history and consanguinity was reported in
the patient's family. The patient was suspected to have EBS based
on the clinical manifestations, and Sanger sequencing was performed
to clarify the diagnosis.
Subjects
The proband, his parents and 100 population-matched
healthy controls (the mean age of the healthy controls was 24 years
old with a range between 18 to 30 years old and a gender ratio of
females/males equal to 1.0) were enrolled in the present study.
Subsequent to obtaining written informed consent from the
participant's mother, peripheral blood samples were collected for
DNA extraction. The present study was approved by the Institutional
Review Board of Xinhua Hospital, Shanghai Jiaotong University
School of Medicine, and was conducted in accordance with the
principles of the Declaration of Helsinki. Ethical approval was
obtained from the Ethics Committee of the Xinhua Hospital
Affiliated to Shanghai Jiao Tong University School of Medicine.
Methods
Genomic DNA was extracted using a TIANamp Blood DNA
kit (Tiangen Biotech Co., Ltd., Beijing, China). Primers flanking
all coding exons and intron-exon boundaries of KRT5 and
KRT14 were designed using Primer Premier version 5.0
(Premier Biosoft, Palo Alto, CA, USA; Table I). Genomic DNA samples were amplified
using polymerase chain reaction (PCR). PCR was performed as
follows: A denaturation step at 94°C for 5 min; 31 cycles of
denaturation at 94°C for 30 sec, an annealing step for 30 sec
(temperature was according to the primers of each fragment), an
extension at 72°C for 1 min and an extension at 72°C for 1 min.
Next, a final extension step was performed at 4°C for 5 min, and
the experiment was repeated 10–20 times. The PCR products were
evaluated by a 2% agarose gel electrophoresis and were further
purified using an AxyPrep DNA Gel Extraction kit (Corning Life
Sciences, Corning, NY, USA), according to the manufacturer's
instructions. Sanger sequencing was subsequently performed using an
ABI PRISM 3730 automated sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Sequencing results were
analyzed by Geneious version 5.6.7 software (Biomatters, Ltd.,
Auckland, New Zealand). An identified mutation was verified in the
corresponding region of the unaffected parents of the proband and
100 population-matched healthy controls. The mutation was described
by comparison with the NCBI cDNA reference sequences NM_000424.3
for KRT5 and NM_000526.4 for KRT14.
 | Table I.List of the primers of the KRT5
and KRT14 genes. |
Table I.
List of the primers of the KRT5
and KRT14 genes.
| Primer name | Primer Sequence | Annealing temperature
(°C) |
|---|
| keratin 5-E01_F |
TGGGTAACAGAGCCACCTTC |
|
| keratin 5-E01_R |
TTGCACAAAGCCAAAACATC | 55 |
| keratin 5-E02_F |
TAGAGGGACGGAAAGAGGTG |
|
| keratin 5-E02_R |
GGAGGTGTCCATGGAAGGTA | 59 |
| keratin
5-E03+4+5_F |
CCCTTCCCACTGCAAAAGTA |
|
| keratin
5-E03+4+5_R |
GAGCCCCATTCTTAGTGTCG | 57 |
| keratin
5-E06+7_F |
AACCAGCCCCACACTATTTG |
|
| keratin
5-E06+7_R |
AGCAGCTTCGCTTTATCAGC | 57 |
| keratin 5-E08_F |
CGAATCATGAGGATGGGAGT |
|
| keratin 5-E08_R |
GGGATGGGAAAAGTTTGGAT | 55 |
| keratin 5-E09_F |
AGGGGGTCCAGTAGAGTGCT |
|
| keratin 5-E09_R |
TTCTGCAATTGGCTTGGTCT | 57 |
| keratin 14-E01_F |
GACAGACATGATGAGGCGGAT |
|
| keratin 14-E01_R |
CTGCCTCCTGTGCTGGAAGG | 65 |
| keratin
14-E02+3_F |
CCTTCCAGCACAGGAGGCAG |
|
| keratin
14-E02+3_R |
CAGCGGATTGGTGTTCCTTAG | 64 |
| keratin
14-E04−8_F |
TGGTGGAACTCCTGACTGTGG |
|
| keratin
14-E04−8_R |
CCATGAACCCCATGACATTG | 60.8 |
The potential impact of an amino acid substitution
on the structure and function of KRT5 and KRT14 proteins was
predicted using the following in silico analysis tools:
PolyPhen2 (http://genetics.bwh.harvard.edu/pph2), SIFT
(http://sift.bii.a-star.edu.sg) and
Mutation Taster (http://www.mutationtaster.org/), which automatically
generated the results following input.
Results
Sequencing results
The results of the present study indicated that
mutation sequencing of KRT14 was negative, whereas a novel
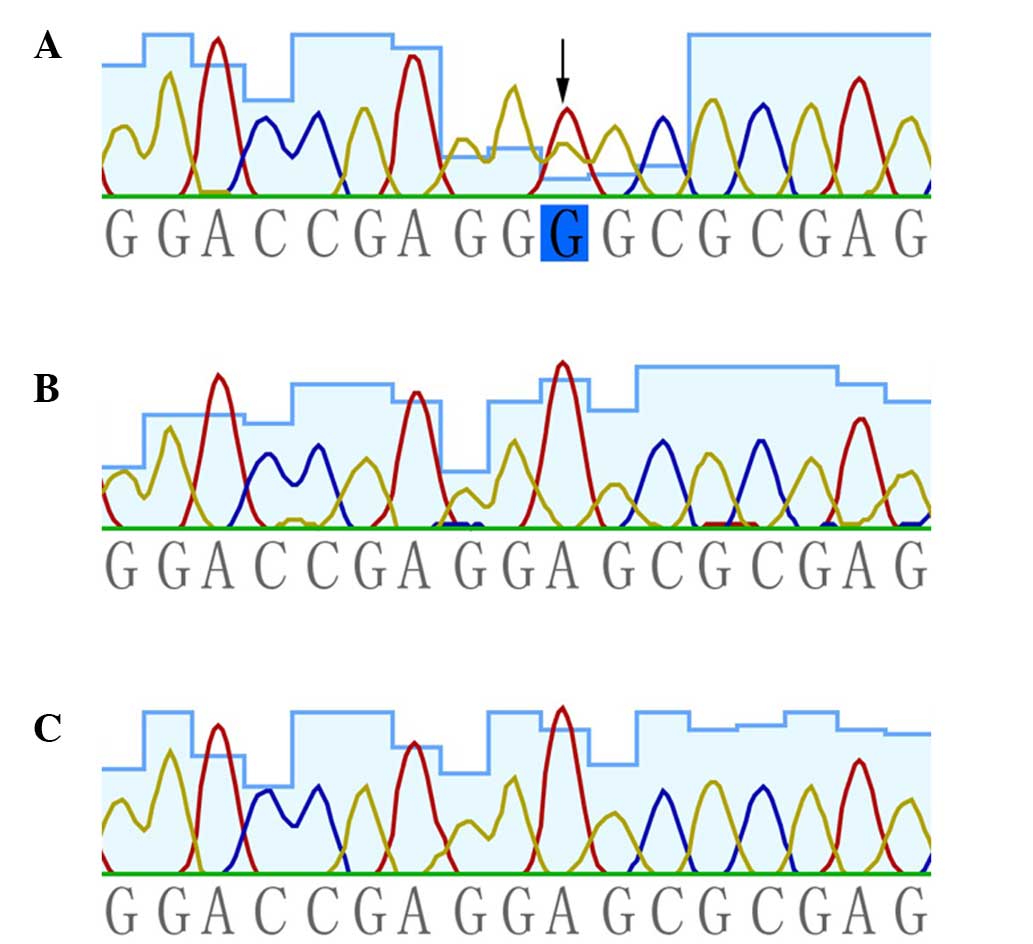
heterozygous missense mutation, c.503C>T (p.Glu168Gly), in
KRT5 was presented in the patient. This mutation was absent
in the patient's unaffected parents and the 100 population-matched
healthy controls (Fig. 2).
Functional consequence
predictions
Analysis using PolyPhen2 indicated that the mutation
c.503A>G (p.Glu168Gly) was ‘probably damaging’. Furthermore,
SIFT and Mutation Taster software predicted the mutation to be
‘deleterious’ and ‘disease causing’, respectively.
Discussion
The locus of the mutation in KRT5 serves an
important role in the phenotype of EBS-gen sev. The majority of
causal variants of EBS-gen sev in KRT5 are missense
mutations that exist in the highly conserved regions of HIP and HTP
(which are critical for the intermediate filament structure and
integrity of the keratin cytoskeleton), and exert a dominant
negative effect on the functional protein structure (2,8,9,14).
Mutations in other regions are typically associated with the milder
subtypes of EBS, namely EBS-loc and EBS-gen intermed (2,8,14).
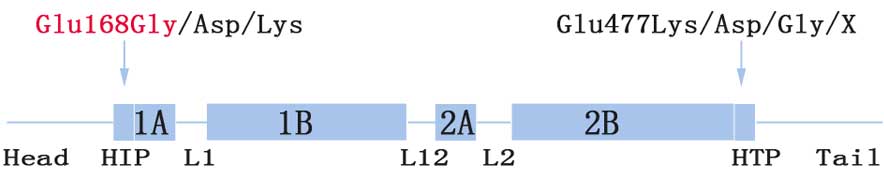
The Glu168 site (codon, GAG) is located at the
boundary between the head and 1A domain (HIP) (Fig. 3). To date, three relevant
substitutions have been reported. More specifically, mutations
c.504G>T (p.Glu168Asp) and c.504G>C (p.Glu168Asp) result in
EBS-gen intermed, while c.502G>A (p.Glu168Lys) is responsible
for EBS-gen sev (15–17). Phenotype severity may be explained by
the fact that Glu and Asp are acidic amino acids that share a
similar structure and polarity, while Lys and Gly are very
different with regards to these features. Contrary to Arg125Cys and
Arg125His in the Arg125 site (a CpG-rich hotspot codon) accounting
for the majority of mutations in KRT14 (8,16,18),
Glu477Lys located at the C-terminal end segments of the helix 2B
domain (HTP) was the most common mutation identified in KRT5
(19). This was more common than the
corresponding region, Glu168 (Fig.
3; 8,14,16–18).
Dowling-Degos disease (DDD; OMIM no. 179850) or
Galli-Galli disease (an acantholytic variant of DDD) are associated
with haploinsufficiency of KRT5 caused by heterozygous
frameshift/nonsense mutations (such as p.Met1?, p.Gln4* and
p.Ile140Asnfs*39) in the head domain of KRT5 (20–22).
Furthermore, p.Pro25Leu (in the head domain) and p.Gly550Alafs*77
(in the tail domain) result in a rare subtype of EBS, known as EBS
with mottled pigmentation (OMIM no. 148040), suggesting that
specific regions in KRT5 may be associated with melanin
transportation and distribution, and malfunction of which can
result in pigmentary phenotypes (20,23).
Although variable phenotypes can arise from distinct
KRT5 mutations, even from an identical mutation in one
pedigree (13), substitutions in the
Glu168 site primarily result in the most severe subtype of EBS,
which is EBS-gen sev. However, there exist exceptions (for
instance, Glu168Asp results in EBS-gen intermed), suggesting that
other factors, such as epigenetic alterations, interchain
interactions of protein structure, modifier genes, environmental
interference and ethnic background, may exert effects that result
in distinct phenotypes.
In the present study, it can be suggested that
Glu168Gly is the pathogenic mutation present in the proband based
on the following: i) Glu168 is highly conserved among different
species (analyzed by the Mutation Taster software; (www.mutationtaster.org/); ii) functional
consequence predictions are deleterious; iii) other pathogenic
mutations in Glu168 have been reported (Fig. 3); and iv) the variant was not present
in the patient's unaffected parents and 100 healthy controls. In
combination with the generalized herpetiform blistering occurring
since birth and improving with age, the 2-year-old male in the
present study was diagnosed with EBS-gen sev.
The patient did not evidently improve after 9 months
from the first time that they appeared at the Department of
Dermatology of Xinhua Hospital (Shanghai, China), and this may be
attributed to the relatively long-term clinical course of EBS-gen
sev, or due to insufficient general management of EBS in the
infant. Furthermore, no treatment was given within these 5 months.
Subsequent general therapy of EBS-gen sev in this patient should
concern the prevention of skin trauma, infection control and the
maintenance of good nutrition. Since a favorable lifelong prognosis
of EBS-gen sev is anticipated following the high mortality rate
period (within a year from birth), prenatal diagnosis and potential
gene therapy will be available to the next generation in the
family.
In conclusion, the current study successfully
confirmed a diagnosis of EBS-gen sev by revealing a novel de
novo heterozygous missense mutation c.503A>G in the HIP of
KRT5, expanding the existing mutation spectrum.
Acknowledgements
The authors would like to thank all subjects for
their ongoing participation in the study. The present study was
supported by grants from the PhD Programs Foundation of the
Ministry of Education of China (grant no. 20130073120014) and the
Natural Science Foundation of Shanghai Jiaotong University School
of Medicine (grant no. 13XJ10023).
References
|
1
|
Fine JD, Bruckner-Tuderman L, Eady RA,
Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A,
Jonkman MF, et al: Inherited epidermolysis bullosa: Updated
recommendations on diagnosis and classification. J Am Acad
Dermatol. 70:1103–1126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coulombe PA, Hutton ME, Vassar R and Fuchs
E: A function for keratins and a common thread among different
types of epidermolysis bullosa simplex diseases. J Cell Biol.
115:1661–1674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coulombe PA, Hutton ME, Letal A, Hebert A,
Paller AS and Fuchs E: Point mutations in human keratin 14 genes of
epidermolysis bullosa simplex patients: Genetic and functional
analyses. Cell. 66:1301–1311. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hiremagalore R, Kubba A, Bansel S and
Jerajani H: Immunofluorescence mapping in inherited epidermolysis
bullosa: A study of 86 cases from India. Br J Dermatol.
172:384–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Intong LR and Murrell DF: Inherited
epidermolysis bullosa: New diagnostic criteria and classification.
Clin Dermatol. 30:70–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fine JD, Johnson LB, Weiner M, Li KP and
Suchindran C: Epidermolysis bullosa and the risk of
life-threatening cancers: The National EB Registry experience,
1986–2006. J Am Acad Dermatol. 60:203–211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sawamura D, Nakano H and Matsuzaki Y:
Overview of epidermolysis bullosa. J Dermatol. 37:214–219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yasukawa K, Sawamura D, Goto M, Nakamura
H, Jung SY, Kim SC and Shimizu H: Epidermolysis bullosa simplex in
Japanese and Korean patients: Genetic studies in 19 cases. Br J
Dermatol. 155:313–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Steinert PM, Marekov LN, Fraser RD and
Parry DA: Keratin intermediate filament structure: Crosslinking
studies yield quantitative information on molecular dimensions and
mechanism of assembly. J Mol Biol. 230:436–452. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li XL, Xiao SX, Peng ZH, Liu Y, Pan M and
Zhou SN: A mutation in exon 1 of keratin 14 resulting in a Chinese
family with epidermolysis bullosa simplex Dowling-Meara. J Eur Acad
Dermatol Venereol. 21:979–981. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yuan H, Liu F, Xiao B, He Y, Liang Y and
Liu J: Mutation screening of entire keratin 5 and keratin 14 genes
and identification of a novel mutation in a Chinese family with
epidermolysis bullosa simplex Dowling-Meara. J Eur Acad Dermatol
Venereol. 22:1510–1512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu JW and Xiao SX: A recurrent keratin 14
mutation in Dowling-Meara epidermolysis bullosa simplex in a
Chinese family. J Eur Acad Dermatol Venereol. 23:484–486. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deng W, Yuan P, Lai W, Chen M, Wang Y and
Dai S: A novel KRT5 mutation, p. Lys199Asn, is associated with
three subtypes of epidermolysis bullosa simplex phenotypes in a
single Chinese family. J Dermatol Sci. 64:241–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang TW, Lee JS, Kim SE, Oh SW and Kim SC:
Novel and recurrent mutations in Keratin 5 and 14 in Korean
patients with Epidermolysis bullosa simplex. J Dermatol Sci.
57:90–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shinkuma S, Nishie W, Jacyk WK, Natsuga K,
Ujiie H, Nakamura H, Akiyama M and Shimizu H: A novel keratin 5
mutation in an african family with epidermolysis bullosa simplex
indicates the importance of the amino acid located at the boundary
site between the H1 and coil 1A domains. Acta Derm Venereol.
93:585–587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müller FB, Küster W, Wodecki K, Almeida H
Jr, Bruckner-Tuderman L, Krieg T, Korge BP and Arin MJ: Novel and
recurrent mutations in keratin KRT5 and KRT14 genes in
epidermolysis bullosa simplex: Implications for disease phenotype
and keratin filament assembly. Hum Mutat. 27:719–720. 2006.
View Article : Google Scholar
|
|
17
|
Murrell DF, Trisnowati N, Miyakis S and
Paller AS: The yin and the yang of keratin amino acid substitutions
and epidermolysis bullosa simplex. J Invest Dermatol.
131:1787–1790. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bolling MC, Lemmink HH, Jansen GH and
Jonkman MF: Mutations in KRT5 and KRT14 cause epidermolysis bullosa
simplex in 75% of the patients. Br J Dermatol. 164:637–644.
2011.PubMed/NCBI
|
|
19
|
Stephens K, Ehrlich P, Weaver M, Le R,
Spencer A and Sybert VP: Primers for exon-specific amplification of
the KRT5 gene: Identification of novel and recurrent mutations in
epidermolysis bullosa simplex patients. J Invest Dermatol.
108:349–353. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Betz RC, Planko L, Eigelshoven S, Hanneken
S, Pasternack SM, Bussow H, Van Den Bogaert K, Wenzel J,
Braun-Falco M, Rutten A, et al: Loss-of-function mutations in the
keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet.
78:510–519. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo L, Luo X, Zhao A, Huang H, Wei Z, Chen
L, Qin S, Shao L, Xuan J, Feng G, et al: A novel heterozygous
nonsense mutation of keratin 5 in a Chinese family with
Dowling-Degos disease. J Eur Acad Dermatol Venereol. 26:908–910.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liao H, Zhao Y, Baty DU, McGrath JA,
Mellerio JE and McLean WH: A heterozygous frameshift mutation in
the V1 domain of keratin 5 in a family with Dowling-Degos disease.
J Invest Dermatol. 127:298–300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Planko L, Böhse K, Höhfeld J, Betz RC,
Hanneken S, Eigelshoven S, Kruse R, Nöthen MM and Magin TM:
Identification of a keratin-associated protein with a putative role
in vesicle transport. Eur J Cell Biol. 86:827–839. 2007. View Article : Google Scholar : PubMed/NCBI
|