Introduction
Atherosclerosis (AS) occurs as a result of
endothelium injury, and leads to clogged arteries, resulting in
heart attacks and strokes (1). It
has recently been demonstrated that oxidized low-density
lipoprotein (ox-LDL) has a key role in early inflammatory processes
and may induce atherosclerotic lesions (2). In the process of atherosclerotic lesion
formation, ox-LDL promotes the initiation of monocyte invasion and
is taken up by monocytes/macrophages and endothelial cells via
scavenger receptors on the cell surface (2). The subsequent accumulation of
cholesterol in macrophages and foam cells is an indicator of
atherosclerotic lesion formation. In addition, ox-LDL increases the
permeability of endothelial cells and promotes their dysfunction
(2). Previous studies have
demonstrated that ox-LDL may significantly delay endothelium wound
healing, and that the expression of numerous key genes in
endothelial cells was changed following ox-LDL treatment, thus
altering the function of the endothelium (3,4).
At the molecular level, ox-LDL has been demonstrated
to promote the expression of adhesion molecules, heat shock
proteins and coagulation proteins; to suppress the production of
endothelium-derived nitric oxide (NO) and prostacyclin; and to
induce the expression of various proinflammatory cytokines and
growth factors in vascular cells (5–9).
Therefore, inhibiting ox-LDL-induced vascular endothelial cell
injury may be a potential therapeutic strategy for AS.
Previous studies have demonstrated that endothelial
dysfunction and inflammation are precursors of AS (10,11).
Numerous pathological conditions, including dyslipidemia,
hypertension and hyperglycemia, have been associated with the
overexpression of reactive oxygen species (ROS), which may
stimulate endothelial cells and induce inflammatory responses
(12,13). The necrosis of injured endothelial
cells may result in the release of various pro-inflammatory
factors, including intercellular adhesion molecule-1 and vascular
cell adhesion molecule-1; these pro-inflammatory factors induce the
formation of atherosclerotic plaques (14). Therefore, anti-inflammatory responses
may be important for the prevention of AS.
Various growth factors and cytokines are involved in
angiogenesis, including vascular endothelial growth factor (VEGF).
VEGF binds to VEGF receptors (VEGFR) on the surface of cells, which
induces the phosphorylation of downstream signaling molecules,
including mitogen-activated protein kinase, focal adhesion kinase,
Src kinase and signal transducer (15). The main subtypes of VEGFRs are VEGFRs
1–3, which are predominantly located on the surface of healthy
tissue cells. However, VEGFRs have been observed to be upregulated
during embryonic and tumor angiogenesis. VEGFR1 is a decoy receptor
that has a low tyrosine kinase activity (16). Conversely, previous studies have
suggested that VEGFR2 has a critical role in cell proliferation,
migration and tube formation, leading to angiogenesis (17,18).
However, to the best of our knowledge, the
cytoprotective effect of VEGFR2 on oxLDL-induced human umbilical
vein endothelial cell (HUVEC) injury has yet to be investigated
clearly. The present study aimed to investigate the effect of
ox-LDL on angiogenesis by exposing HUVECs to ox-LDL and performing
endothelial proliferation and angiogenesis assays. In addition, the
role of VEGFR2 in AS was determined.
Materials and methods
Chemicals, antibodies and
reagents
LDL was purchased from R&D Systems GmbH
(Wiesbaden, Germany). Calcein-AM was purchased from Dojindo
Molecular Technologies, Inc. (Shanghai, China). Rabbit anti-VEGFR2
polyclonal antibody was obtained from Abcam (1:100; ab39256;
Cambridge, UK). Phycoerythrin (PE)-conjugated mouse anti-VEGFR2
monoclonal antibody (1:500; FAB357P; clone 12G5) and purified mouse
immunoglobulin (Ig)G (1:500; 550874) were purchased from BD
Biosciences (Heidelberg, Germany). Recombinant human VEGF165
(11066-HNAB-500) was obtained from Sino Biological, Inc. (Beijing,
China). Rabbit anti-β-actin (1:500; ab16039, Abcam) and rabbit
anti-human glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:500;
SAB2108266) polyclonal antibodies were obtained from Sigma-Aldrich
Chemie GmbH (Deisenhofen, Germany).
Cell culture and LDL treatment
In total 2 × 105 HUVECs (AllCells, LLC,
Shanghai, China) were cultured for 3–5 passages in Dulbecco's
modified Eagle's medium (DMEM; PromoCell GmbH, Heidelberg, Germany)
supplemented with 10% fetal bovine serum (unless otherwise
specified), 0.1 ng/ml human epidermal growth factor, 1 ng/ml basic
fibroblast growth factor (bFGF), 90 ng/ml heparin and 1 ng/ml
hydrocortisone (all purchased from Sigma-Aldrich Chemie GmbH).
Cells were incubated at 37°C for 24 h in a humidified 5%
CO2 incubator at 21% O2. Ox-LDL was prepared
by exposure of native LDL to 5 µM copper sulphate (Sigma-Aldrich
Chemie GmbH) at 37°C for 3 h. The reaction was stopped by the
addition of 0.25 mM ethylenediaminetetraacetic acid, as described
previously (19). Ox-LDL (0–100
µg/ml) was added to the cells alone or in combination with each
other at 37°C for 24 h.
Cell proliferation assay
The MTT assay (Sigma-Aldrich Chemie GmbH) was used
to assess the proliferation of HUVECs, according to the
manufacturer's protocol. Briefly, HUVECs (1×104
cells/well) were plated onto 96-well plates and treated with ox-LDL
at 37°C for 24 h, after which MTT (20 µl, 5 g/l) was added to media
and incubated for 4 h. Subsequently, the supernatant was removed by
centrifugation at 1000xg for 10 min at 4°C, and dimethylsulfoxide
was added to solubilize the formazan crystals. Absorbance was
measured at 560 nm using an ELISA plate reader.
Cell apoptosis assay
The apoptosis of HUVECs induced by ox-LDL starvation
was detected by Annexin V-fluorescein isothiocyanate (FITC)
staining assays (Nanjing KeyGen Biotech, Co., Ltd., Nanjing,
China). Briefly, 1×104 HUVECs were cultured with DMEM
supplemented with 10 ng/ml VEGF and in the presence or absence of
ox-LDL for 48 h. The cells were incubated with ox-LDL (0–100
µg/ml), VEGF (10 ng/ml) or both for 48 h. HUVECs were then
collected and washed with phosphate-buffered saline (PBS) three
times, after which Annexin V-FITC and propidium iodide were added
to the washed cells (106 cells/ml) for 15 min at room
temperature in the dark. Subsequently, fluorescence-activated cell
sorting buffer was added and the cells were immediately analyzed by
flow cytometry.
Endothelial cell wound healing
assay
HUVECs (1×105 cells/well) were seeded
into 12-well plates and were scratched with pipette tips, followed
by treatment with 100 µg/ml ox-LDL. Migration of cells into the
wound was then observed at 37°C for 24 h. The migrated cells were
visualized and imaged under a microscope at various time points.
Experiments were performed in triplicate at least three times.
Tube formation assay
HUVECs (2×104) were seeded into
Matrigel-coated wells of a 96-well plate. The cells were incubated
with ox-LDL (0–100 µg/ml) and/or VEGF (10 ng/ml) for 24 h, during
which cells were maintained in an incubator at 37°C containing 21%
O2. Images were captured at low magnification
(magnification, ×5) under a microscope and tubes were counted. Only
perfectly continuous tubes between two branching points were
included. For each condition, three independent experiments were
performed, of which mean tube numbers are presented.
Intracellular ROS production
HUVECs (2×105 cells/well) were seeded
into a 6-well plate and treated with ox-LDL (0–100 µg/ml) for 24 h.
The dichlorofluorescein diacetate (DCFH-DA) assay (Sigma-Aldrich
Chemie GmbH) was used to measure the intracellular levels of ROS.
Briefly, following treatment, the cells were incubated with 10 µM
DCFH-DA for 30 min at 37°C. Flow cytometry was used to detect the
fluorescence intensity.
Measurement of caspase-3 activity
The activity of caspase-3 was measured using a
Caspase-3 Cellular Activity Assay kit (Nanjing KeyGen Biotech, Co.,
Ltd.), according to the manufacturer's protocol. Briefly,
1×104 HUVECs were removed from culture dishes, washed
twice with PBS and centrifuged at 10,000 × g for 1 min at 4°C. Cell
pellets were then treated for 30 min with ice-cold lysis buffer
provided by the manufacturer of the kit. Cell suspensions were then
centrifuged at 10,000 × g for 1 min at 4°C and the supernatants
were transferred to a clear tube. To each tube, 2X reaction buffer
and specific substrate for caspase-3 were added, and the tubes were
incubated at 37°C for 4 h. Following incubation, caspase-3 activity
was measured at 405 nm using a micrometer plate reader.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
A total of 1×104 HUVECs were treated with
various concentrations of ox-LDL (0–100 µg/ml) for 24 h or 100
µg/ml ox-LDL for 24–72 h, after which total RNA was extracted from
HUVECs using the RNeasy Midi kit (Qiagen GmbH, Hilden, Germany),
according to manufacturer's protocol. Total RNA (1 µl) was reverse
transcribed into cDNA using iScript cDNA synthesis kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). qPCR was performed using
Power SYBR® Green PCR Master Mix (Eurogentec, Verviers,
Belgium) on the StepOnePlus™ Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
following primers were used: VEGFR2 forward,
5′-CTCTTGGCCGTGGTGCCTTTG and reverse, 3′-GTGTGTTGCTCCTTCTTTCAAC;
and GAPDH forward, 5′-CATCTTCCAGGAGCGAGATCC and reverse,
3′-GGTGCAGGTGGCATTGCTGATG. GAPDH was used as housekeeping gene. The
PCR cycling conditions were as follows: Denaturation at 95°C for 10
min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min.
All samples and standards were conducted in triplicate. The
relative mRNA expression was determined using the 2∆∆cq
method. A negative control without RT enzyme and an RT-minus
control (without reverse transcriptase added to the cDNA synthesis
reaction) were used.
Western blot analysis
A total of 2×105 HUVECs were treated with
ox-LDL (0–100 µg/ml) at 37°C for 72 h, after which HUVECs were
washed with PBS and lysed using radioimmunoprecipitation assay
(Thermo Fisher Scientific, Inc.) lysis buffer. Cell lysates were
centrifuged at 10,000 × g for 10 min at 4°C, after which
protein concentrations were determined using a Pierce BCA Protein
Assay kit (23225; Sigma-Aldrich Chemie GmbH). Total lysate proteins
(40 µg) were resuspended in loading buffer and separated by 10%
SDS-PAGE, followed by transfer onto a polyvinylidene difluoride
membrane. For detection of VEGFR2, the membranes were incubated
overnight at 4°C with rabbit anti-VEGFR2 and rabbit anti-β-actin
polyclonal antibodies (1:400). After washing three times with
Tris-buffered saline containing Tween-20, the membranes were
blocked with 5% bovine serum albumin for 1 h at room temperature
and incubated with peroxidase-conjugated goat anti-rabbit IgG
(1:2,000; Abcam) for 1 h and then detected by incubation with
chromomeric substrate, 3, 3′-diaminobenzidine.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparisons among groups were performed by one-way analysis of
variance followed by Tukey's post-hoc test. Comparisons between two
groups were performed by two-tailed unpaired t-tests. Statistical
analysis was performed using SPSS 10.0 software for Windows (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
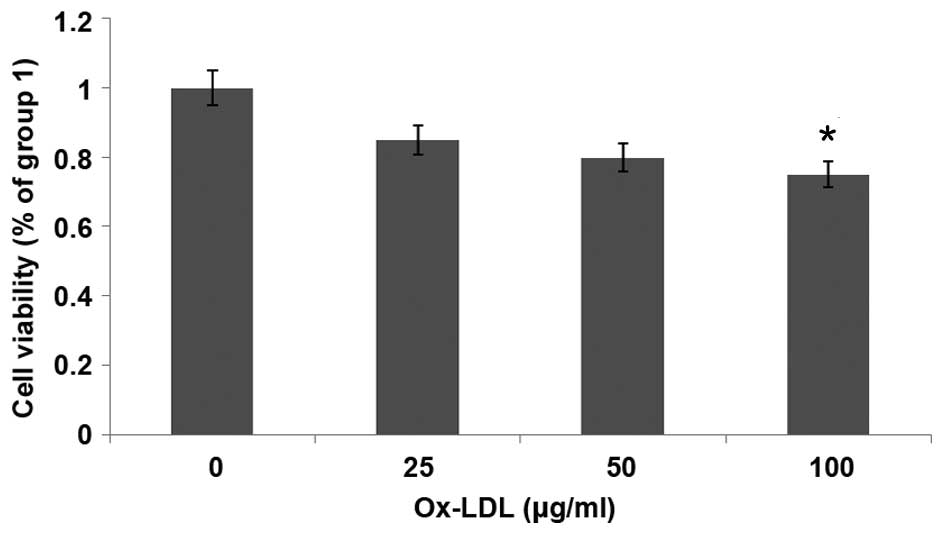
Ox-LDL reduces cell viability in a
dose-dependent manner
MTT assays were used to evaluate the effect of
ox-LDL on the viability of HUVECs. HUVECs were cultured in growth
factor-deprived DMEM containing ox-LDL (0–100 µg/ml) for 24 h.
Fig. 1 shows that HUVEC
proliferation was decreased following treatment with ox-LDL in a
dose-dependent manner; the proliferation of HUVECs was
significantly decreased following treatment with 100 µg/ml ox-LDL
(P<0.05). These results suggest that ox-LDL reduces the
viability of HUVECs.
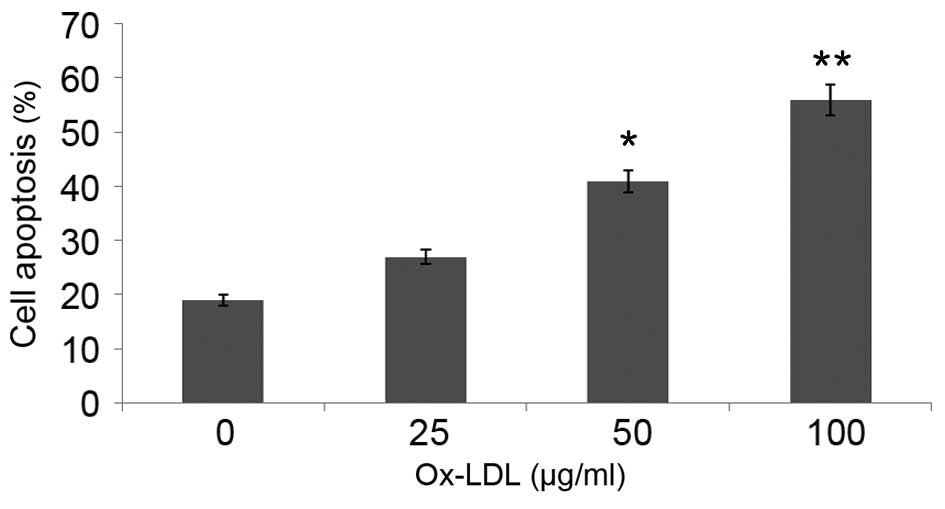
Ox-LDL induces HUVEC apoptosis in a
dose-dependent manner
In order to investigate the effect of ox-LDL on
HUVEC apoptosis, serum deprivation-induced apoptosis of HUVECs was
assessed by flow cytometry. Serum deprivation induced apoptosis of
~19% of HUVECs, which was significantly increased to 56% following
treatment with 100 µg/ml ox-LDL for 48 h (P<0.01; Fig. 2). These results suggest that ox-LDL
induces the apoptosis of HUVECs.
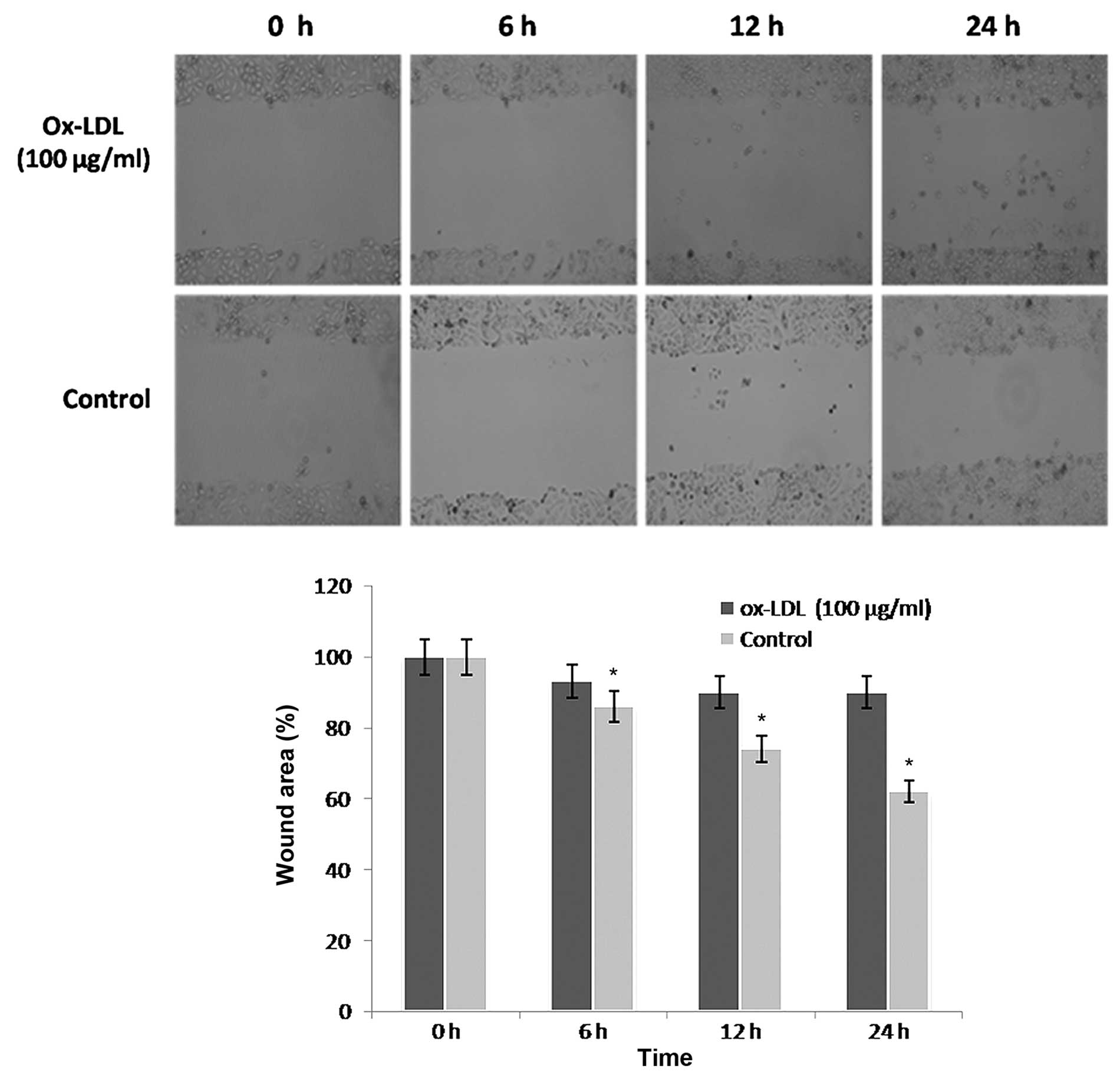
Ox-LDL dose-dependently decreases
HUVEC migration
Cell migration is an essential process in
angiogenesis. The present study performed wound healing assays to
investigate the effects of ox-LDL on the migration of HUVECs.
ox-LDL (100 µg/ml) markedly inhibited the migration of HUVECs into
the wound area (Fig. 3). This effect
was significant at 6, 12 and 24 h, as compared with the control
(P<0.05; Fig. 3).
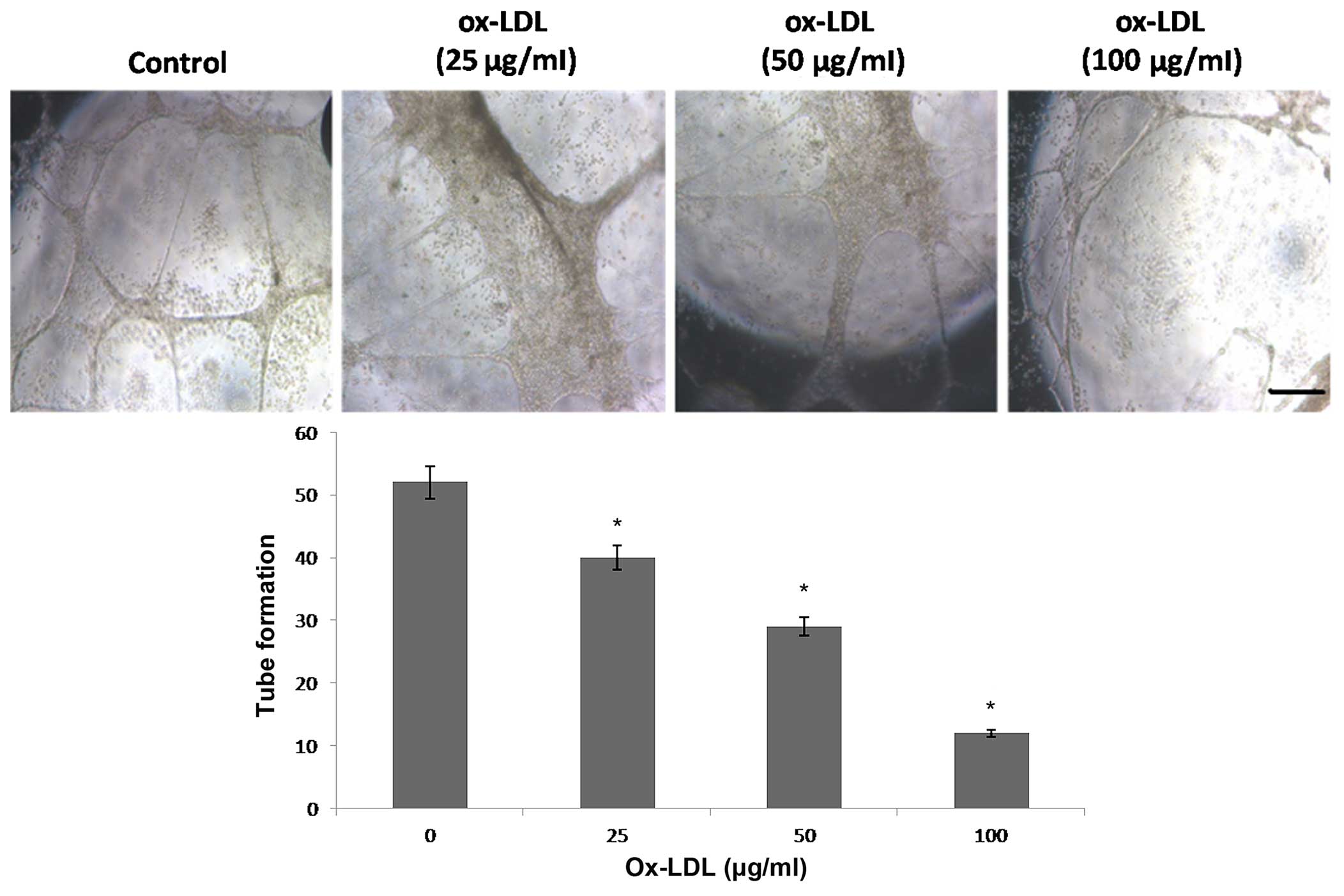
Ox-LDL dose-dependently inhibits
angiogenesis
In order to elucidate the potential underlying
mechanisms of the effects of ox-LDL on angiogenesis, the
tube-forming ability of HUVECs was assessed in vitro. HUVECs
were cultured in DMEM containing 10 ng/ml VEGF and/or ox-LDL (0–100
µg/ml) for 24 h, after which the cells were seeded into
matrigel-coated plates and the lengths of tube-like structures were
measured. VEGF (10 ng/ml) induced HUVEC tube formation. Conversely,
ox-LDL significantly inhibited tube formation by HUVECs in a
dose-dependent manner (Fig. 4).
These results suggest that ox-LDL is able to suppress HUVEC
angiogenesis in vitro.
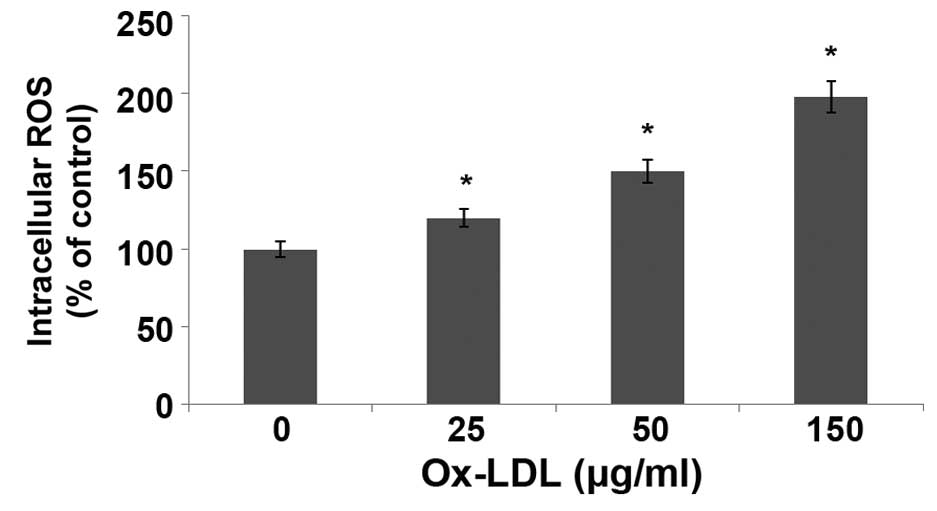
ox-LDL induces overproduction of ROS
in HUVECs
The levels of ROS in HUVECs treated with ox-LDL for
24 h were measured using DCFH-DA staining and flow cytometry. As
shown in Fig. 5, the levels of ROS
were significantly increased in HUVECs following treatment with
ox-LDL in a dose-dependent manner, as compared with the control
(P<0.05). In particular, treatment with 150 µg/ml ox-LDL
increased the levels of ROS in HUVECs by 2.18-fold.
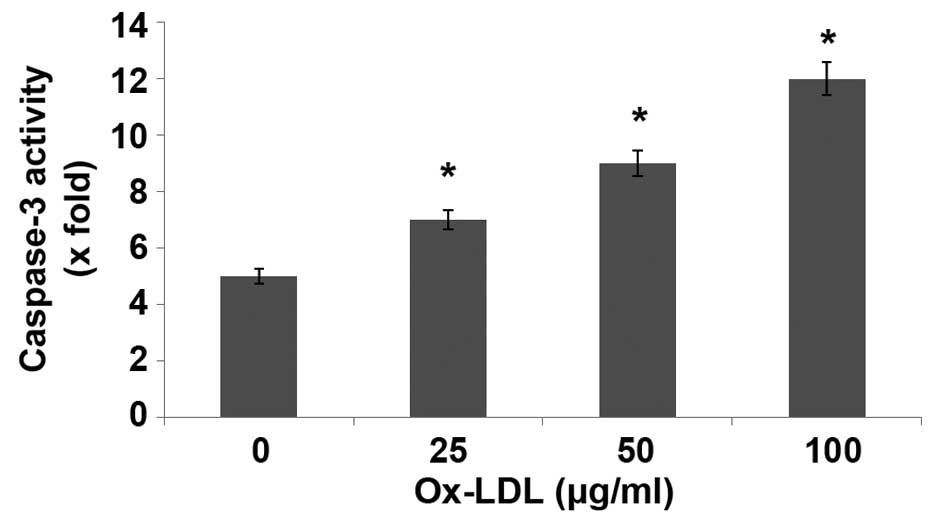
ox-LDL activates caspase-3 in
HUVECs
The present study demonstrated that ox-LDL induced
HUVEC injury, indicating a potential suppressive effect on HUVEC
angiogenesis. Caspases are crucial mediators of programmed cell
death (apoptosi). Among them, caspase-3 is a frequently activated
death protease, catalyzing the specific cleavage of many key
cellular proteins. Incubation of HUVECs with ox-LDL for 24 h
significantly increased caspase-3 activity, as compared with the
control (P<0.05; Fig. 6).
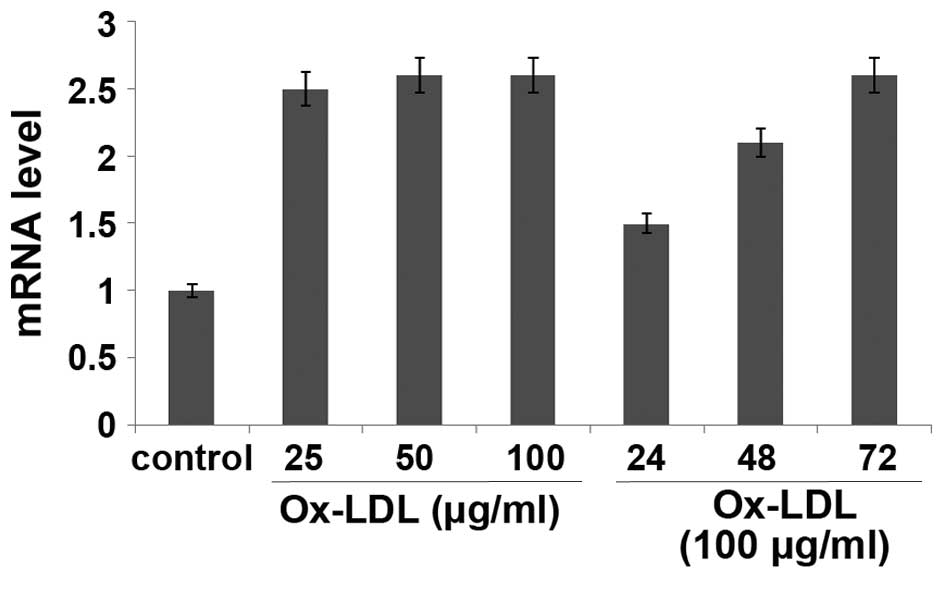
Ox-LDL regulates VEGFR2 expression at
the post transcriptional level
The present study demonstrated that ox-LDL inhibited
VEGF-induced angiogenesis of HUVECs. In order to investigate the
underlying mechanisms, the mRNA and protein expression levels of
VEGFR2 in ox-LDL-treated HUVECs were determined by RT-qPCR and
western blotting, respectively. The mRNA expression levels of
VEGFR2 were increased in HUVECs treated with various concentrations
of ox-LDL for 24 h or with 100 µg/ml ox-LDL for various durations;
however, the difference was not significant, as compared with the
control (P>0.05; Fig. 7).
Conversely, the protein expression of VEGFR was markedly decreased
in HUVECs treated with ox-LDL, as compared with the control
(Fig. 8). These results suggest that
ox-LDL regulates VEGFR2 expression at the protein, but not the mRNA
level.
Discussion
Epidemiological studies have examined the incidence
of AS being 79.9% in Chinese people >60 years old. AS is serious
but is not yet a global health emergency. Ox-LDL-induced vascular
endothelial damage has been demonstrated to be a driving force in
the initiation and development of AS (20). A key therapeutic strategy for AS is
the promotion of angiogenesis. Previous studies have used various
strategies to induce angiogenesis, including the delivery of VEGF
(21), viral vectors (22–24) or
plasmids (25,26); however, few have shown success, which
may be due to the fact that the molecular mechanisms underlying
angiogenesis are largely unknown. Using proliferation, migration
and tube formation assays, the present study demonstrated that
ox-LDL impaired angiogenesis by HUVECs in vitro. Previous
studies have associated hypercholesterolemia with impaired
angiogenesis, and hypercholesterolemia has been shown to enhance
oxidative stress resulting in impaired inflammation in vivo
(27–30). In the present study, ox-LDL impaired
the ability of HUVECs to undergo angiogenesis by decreasing the
viability and migratory and tube-forming abilities of the cells.
Therefore, the present study provides novel insights into the
effects of hypercholesterolemia on angiogenesis.
In the present study, ox-LDL exposure increased ROS
production and caspase-3 activity in HUVECs. In addition, ox-LDL
was observed to induce the apoptosis of HUVECs. As an executioner
caspase, caspase-3 exhibits negligible activity until it is cleaved
by an initiator caspase following induction of cell apoptotic
events. Therefore, under normal conditions, healthy cells express
full length, inactive caspase-3. As is shown in the present study,
exposure of HUVECs to ox-LDL increased caspase-3 activity, thus
suggesting that the rate of apoptosis was increased. These results
support the hypothesis that ox-LDL is able to induce the apoptosis
of HUVECs. This is significant since atherosclerosis has previously
been associated with progressive endothelial cell loss (31). Therefore, the present study provides
a potential mechanism by which ox-LDL exposure may enhance
oxidative injury, in particular via induction of ROS production and
activation of caspase-3.
Increasingly, it has been suggested that VEGFR2
drives the angiogenic response. VEGFR2 abundance and activation of
the downstream signaling pathway in endothelial cells has been
reported to be decreased under hypoxic conditions, such as those
encountered in coronary heart disease, peripheral occlusive artery
disease and ischemic stroke; and this was consistent with the
results of the present study. In addition, previous studies
demonstrated that ox-LDL impaired endothelial cell proliferation
and migration via decreasing bFGF expression (32) and activating NO synthase/Akt
signaling pathway (33).
Furthermore, it has previously been shown that ox-LDL exposure
decreased the expression of VEGFR1 in human macrophages (34), and internalized, ubiquinated and
proteolytically degraded forms of VEGFR1 have been detected
following ox-LDL exposure. However, whether ox-LDL affects the role
of VEGFR2 in the angiogenic response pathway has yet to be
elucidated.
The ox-LDL-mediated increase in oxidative
stress-induced damage of HUVECs is suspected to underlie the
pathogenesis of AS. The present study aimed to investigate the
effects of ox-LDL on HUVEC apoptosis and the underlying mechanisms.
Using endothelial proliferation and tube formation assays, the
present study provided new evidence that ox-LDL exposure markedly
affected HUVEC angiogenesis. The western blotting data confirmed
that VEGFR2 was degraded following ox-LDL exposure, and this
decrease in VEGFR2 expression may have inhibited angiogenesis by
limiting the activation of signaling pathways downstream of VEGFR2.
The results demonstrated that VEGFR2 receptor function was
significantly affected following ox-LDL exposure.
A limitation of the present study was that only the
in vitro effects of ox-LDL on HUVECs were analyzed; the
dose-dependent association between ox-LDL and HUVEC response may be
more complex in vivo. Further studies are required to assess
the effect of ox-LDL on vessel formation in animal models, and to
develop therapeutic strategies for repairing angiogenesis under
hypoxic conditions.
In conclusion, the present study aimed to
investigate the molecular mechanisms underlying the involvement of
VEGFR2 in the regulation of oxidative stress and HUVEC injury in
AS. The results of the present study suggested that ox-LDL was able
to alter endothelial cell survival and function, and that
downregulation of VEGFR2 expression may underlie the development of
AS.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundation of China (no. 81401870) and the Shanghai
Municipal Science and Technology Commission (no. 13ZR1459000).
References
|
1
|
Mannarino E and Pirro M: Molecular biology
of atherosclerosis. Clin Cases Miner Bone Metab. 5:57–62.
2008.PubMed/NCBI
|
|
2
|
Mitra S, Deshmukh A, Sachdeva R, Lu J and
Mehta JL: Oxidized low-density lipoprotein and atherosclerosis
implications in antioxidant therapy. Am J Med Sci. 342:135–142.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kolluru GK, Bir SC and Kevil CG:
Endothelial dysfunction and diabetes: effects on angiogenesis,
vascular remodeling, and wound healing. Int J Vasc Med. Feb
12–2012.(Epub ahead of print) doi: 10.1155/2012/918267. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mathieu P, Pibarot P and Després JP:
Metabolic syndrome: The danger signal in atherosclerosis. Vasc
Health Risk Manag. 2:285–302. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen CH, Jiang W, Via DP, Luo S, Li TR,
Lee YT and Henry PD: Oxidized low-density lipoproteins inhibit
endothelial cell proliferation by suppressing basic fibroblast
growth factor expression. Circulation. 101:171–177. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue M, Itoh H, Tanaka T, Chun TH, Doi K,
Fukunaga Y, Sawada N, Yamshita J, Masatsugu K, Saito T, et al:
Oxidized LDL regulates vascular endothelial growth factor
expression in human macrophages and endothelial cells through
activation of peroxisome proliferator-activated receptor-gamma.
Arterioscler Thromb Vasc Biol. 21:560–566. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuzuya M, Ramos MA, Kanda S, Koike T, Asai
T, Maeda K, Shitara K, Shibuya M and Iguchi A: VEGF protects
against oxidized LDL toxicity to endothelial cells by an
intracellular glutathione-dependent mechanism through the KDR
receptor. Arterioscler Thromb Vasc Biol. 21:765–770. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jovinge S, Ares MP, Kallin B and Nilsson
J: Human monocytes/macrophages release TNF-alpha in response to
Ox-LDL. Arterioscler Thromb Vasc Biol. 16:1573–1579. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Erl W, Weber PC and Weber C: Monocytic
cell adhesion to endothelial cells stimulated by oxidized low
density lipoprotein is mediated by distinct endothelial ligands.
Atherosclerosis. 136:297–303. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Antoniades C, Demosthenous M, Tousoulis D,
Antonopoulos AS, Vlachopoulos C, Toutouza M, Marinou K, Bakogiannis
C, Mavragani K, Lazaros G, et al: Role of asymmetrical
dimethylarginine in inflammation-induced endothelial dysfunction in
human atherosclerosis. Hypertension. 58:93–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thorin-Trescases N, Voghel G, Gendron ME,
Krummen S, Farhat N, Drouin A, Perrault LP and Thorin E:
Pathological aging of the vascular endothelium: Are endothelial
progenitor cells the sentinels of the cardiovascular system? Can J
Cardiol. 21:1019–1024. 2005.PubMed/NCBI
|
|
12
|
Chen YH, Lin SJ, Chen YL, Liu PL and Chen
JW: Anti-inflammatory effects of different drugs/agents with
antioxidant property on endothelial expression of adhesion
molecules. Cardiovasc Hematol Disord Drug Targets. 6:279–304. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Alcaide P, Liu L, Sun J, He A,
Luscinskas FW and Shi GP: Regulation of endothelial cell adhesion
molecule expression by mast cells, macrophages, and neutrophils.
PLoS One. 6:e145252011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chi Z and Melendez AJ: Role of cell
adhesion molecules and immune-cell migration in the initiation,
onset and development of atherosclerosis. Cell Adh Migr. 1:171–175.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerhardt H, Golding M, Fruttiger M,
Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C,
Alitalo K, Shima D and Betsholtz C: VEGF guides angiogenic
sprouting utilizing endothelial tip cell filopodia. J Cell Biol.
161:1163–1177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen B, Zhao J, Zhang S, Wu W and Qi R:
Aspirin inhibits the production of reactive oxygen species by
downregulating Nox4 and inducible nitric oxide synthase in human
endothelial cells exposed to oxidized low-density lipoprotein. J
Cardiovasc Pharmacol. 59:405–412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pirillo A, Norata GD and Catapano AL:
LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. Jul
10–2013.(Epub ahead of print) doi: 10.1155/2013/152786. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Henry TD, Annex BH, McKendall GR, Azrin
MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman
DS, et al: VIVA Investigators: The VIVA trial: Vascular endothelial
growth factor in Ischemia for Vascular Angiogenesis. Circulation.
107:1359–1365. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mäkinen K, Manninen H, Hedman M, Matsi P,
Mussalo H, Alhava E and Ylä-Herttuala S: Increased vascularity
detected by digital subtraction angiography after VEGF gene
transfer to human lower limb artery: A randomized,
placebo-controlled, double-blinded phase II study. Mol Ther.
6:127–133. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hedman M, Hartikainen J, Syvänne M,
Stjernvall J, Hedman A, Kivelä A, Vanninen E, Mussalo H, Kauppila
E, Simula S, et al: Safety and feasibility of catheter-based local
intracoronary vascular endothelial growth factor gene transfer in
the prevention of postangioplasty and in-stent restenosis and in
the treatment of chronic myocardial ischemia: Phase II results of
the Kuopio Angiogenesis Trial (KAT). Circulation. 107:2677–2683.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rajagopalan S, Mohler ER III, Lederman RJ,
Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler
PD, Rasmussen HS and Annex BH: Regional angiogenesis with vascular
endothelial growth factor in peripheral arterial disease: A phase
II randomized, double-blind, controlled study of adenoviral
delivery of vascular endothelial growth factor 121 in patients with
disabling intermittent claudication. Circulation. 108:1933–1938.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kukuła K, Chojnowska L, Dąbrowski M,
Witkowski A, Chmielak Z, Skwarek M, Kądziela J, Teresińska A,
Małecki M, Janik P, et al: Intramyocardial plasmid-encoding human
vascular endothelial growth factor A165/basic fibroblast growth
factor therapy using percutaneous transcatheter approach in
patients with refractory coronary artery disease (VIF-CAD). Am
Heart J. 161:581–589. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kastrup J, Jørgensen E, Rück A, Tägil K,
Glogar D, Ruzyllo W, Bøtker HE, Dudek D, Drvota V, Hesse B, et al:
Euroinject One Group: Direct intramyocardial plasmid vascular
endothelial growth factor-A165 gene therapy in patients with stable
severe angina pectoris A randomized double-blind placebo-controlled
study: The Euroinject One trial. J Am Coll Cardiol. 45:982–988.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duan J, Murohara T, Ikeda H, Katoh A,
Shintani S, Sasaki K, Kawata H, Yamamoto N and Imaizumi T:
Hypercholesterolemia inhibits angiogenesis in response to hindlimb
ischemia: Nitric oxide-dependent mechanism. Circulation. 102:(Suppl
3). III370–III376. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matter CM, Ma L, von Lukowicz T, Meier P,
Lohmann C, Zhang D, Kilic U, Hofmann E, Ha SW, Hersberger M, et al:
Increased balloon-induced inflammation, proliferation, and
neointima formation in apolipoprotein E (ApoE) knockout mice.
Stroke. 37:2625–2632. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Osto E, Matter CM, Kouroedov A, Malinski
T, Bachschmid M, Camici GG, Kilic U, Stallmach T, Boren J, Iliceto
S, et al: c-Jun N-terminal kinase 2 deficiency protects against
hypercholesterolemia-induced endothelial dysfunction and oxidative
stress. Circulation. 118:2073–2080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
ElAli A, Doeppner TR, Zechariah A and
Hermann DM: Increased blood-brain barrier permeability and brain
edema after focal cerebral ischemia induced by hyperlipidemia: Role
of lipid peroxidation and calpain-1/2, matrix
metalloproteinase-2/9, and RhoA overactivation. Stroke.
42:3238–3244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Du F, Zhou J, Gong R, Huang X, Pansuria M,
Virtue A, Li X, Wang H and Yang XF: Endothelial progenitor cells in
atherosclerosis. Front Biosci (Landmark Ed). 17:2327–2349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang PY, Luo S, Jiang T, Lee YT, Lu SC,
Henry PD and Chen CH: Oxidized low-density lipoprotein
downregulates endothelial basic fibroblast growth factor through a
pertussis toxin-sensitive G-protein pathway: Mediator role of
platelet-activating factor-like phospholipids. Circulation.
104:588–593. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chavakis E, Dernbach E, Hermann C, Mondorf
UF, Zeiher AM and Dimmeler S: Oxidized LDL inhibits vascular
endothelial growth factor-induced endothelial cell migration by an
inhibitory effect on the Akt/endothelial nitric oxide synthase
pathway. Circulation. 103:2102–2107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salomonsson L, Svensson L, Pettersson S,
Wiklund O and Ohlsson BG: Oxidised LDL decreases VEGFR-1 expression
in human monocyte-derived macrophages. Atherosclerosis.
169:259–267. 2003. View Article : Google Scholar : PubMed/NCBI
|