Introduction
Traumatic brain injury (TBI) is a common cause of
mortality and disability in adults worldwide (1,2).
Neurological dysfunction and cell death due to TBI are a result of
primary injury associated with the direct physical disruptions of
various pathways or tissues, as well as secondary injury associated
with delayed biochemical changes that are induced by trauma
(3,4). A series of progressive physiological
and pathological changes result in severe secondary injury in TBI
patients. Oxidative stress is widely known to serve a significant
role in the pathogenesis of TBI (5,6), which
causes the impairment of cognition, motor function and neurological
behavior. However, the complicated molecular and cellular
mechanisms underlying TBI remain poorly understood.
A major component of TBI is diffuse axonal injury
(DAI), which refers to the manifestation of microstructural
cellular trauma and various resulting neurochemical reactions,
leading to secondary neuronal death (7). The transportation of axoplasm depends
on the normal formation of cytoskeleton proteins. Protein
carbonylation is commonly observed in cells exposed to oxidants,
resulting in protein aggregation and dysfunction (8), which may precede cellular senescence
and cell death. In addition, protein carbonylation is an important
event in the context of proteostasis due to its non-enzymatic
nature, frequent occurrence and irreversible effects (9). It is well known that the normal
transport of axoplasm relies on the normal cytoskeleton; however,
the association between the axonal injury and protein carbonylation
is not fully understood. Previous studies revealed that the
cytoskeleton includes β-actin, β-tubulin and neurofilaments, which
are the principal target proteins of carbonylation in neurological
disease (10,11). As disease progresses from the
inflammatory to the neurodegenerative phase, an inappropriate
removal of oxidized cytoskeletal proteins may occur (11). Furthermore, a previous study
demonstrated that the levels of carbonyl proteins were increased in
TBI rats (12). Actin is involved in
the manifold cellular processes, and is thus a sensitive target
protein to this oxidative modification (8). The increase in the actin content of
carbonyl groups detected in vivo indicates drastic oxidative
modification leading to significant functional impairments
(13).
The aim of present study was to investigate the
effectiveness of inhibiting carbonylation of cytoskeletal proteins
in regulating oxidative damage in TBI. The protein carbonylation of
two cytoskeletal proteins, β-actin and β-tubulin, was detected
following the exposure of PC12 cell lines to
H2O2. Furthermore, the carboxylation of these
two cytoskeletal proteins was measured after pretreatment of the
PC12 cell lines with aminoguanidine (AG), as well as overexpression
of proteasome, in order to compare and provide the underlying
mechanisms of cytoskeletal protein carbonylation mediating the
development of brain injury following trauma.
Materials and methods
Antibodies and reagents
Purified β-actin, β-tubulin and cytoskeletonal
protein aggregation kits were purchased from Cytoskeleton, Inc.
(Denver, CO, USA). Dulbecco's Modified Eagle's medium (DMEM) and
Lipofectamine 2000 reagent were obtained from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Anti-β-actin monoclonal
antibody was commercially available from Genetex, Inc. (cat. no.
GTX109639; Irvine, CA, USA). Anti-β-tubulin antibody (cat. no.
T4026) and phenylmethylsulfonyl fluoride (PMSF) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). The peroxidase-conjugated
affinity goat anti-mouse IgG secondary antibody was obtained from
Roche Diagnostics (Basel, Switzerland). An expression vector
pCMV-HA (cat. no. Trans 18–35; Clontech; Takara Bio Inc., Mountain
View, CA, USA) encoding for the full-length β5 subunit cDNA used
for transfection was constructed by SyngenTech Co., Ltd. (Shanghai,
China). The enhanced chemiluminescence (ECL) assay kit (cat. no.
PA112) used in western blotting was commercially available from
Tiangen Biotech Co., Ltd. (Shanghai, China).
Cell culture and oxidative stress
induction
PC12 cells (American Type Culture Collection,
Rockville, MD, USA) were routinely cultured in DMEM supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
in a 37°C incubator with an atmosphere of 5% CO2. PC12
cells were subjected to oxidative stress by treatment with various
concentrations of H2O2 (100, 200 and 300
µM) for 48 h, or with 300 µM H2O2
for different time durations (24 or 48 h). PC12 cells in
the control group were treated with culture medium only.
Levels of glutathione (GSH) and
thiobarbituric acid reactive substances (TBARS)
GSH is one of the most important factors protecting
from oxidative attacks by reactive oxygen species, since it
functions as a reducing agent and free-radical trapper (14). In addition, TBARS is a marker of
lipid peroxidation and oxidative damage (15). Therefore, the present study
investigated the levels of GSH and TBARS in PC12 cells that were
subjected to H2O2-induced oxidative stress in
order to determine the effect of oxidative stress in these
cells.
For TBARS detection, 100 µl plasma were incubated
with 500 µl Tris-HCl (Amresco Inc., Solon, OH, USA) and 500 µl 35%
trichloroacetic acid (TCA; Merck KGaA, Darmstadt, Germany) for 10
min at room temperature. Next, the samples were incubated with 2 M
Na2SO4 and 55 mM TBA solution at 95°C for 45
min, and then cooled on ice for 5 min. Subsequent to treatment with
70% TCA, the samples were centrifuged at 12,000 × g for 3
min. The TBARS product was measured at 530 nm using a
spectrophotometer (Ultrospec 500 Pro; GE Healthcare Life Sciences,
Chicago, IL, USA).
For the detection of GSH, 5% TCA, 67 mM
sodium/potassium phosphate buffer and 1 mM
5,5′-dithiobis(2-nitrobenzoic acid) (Vicmed, Xuzhou, China) were
mixed with 20 µl erythrocyte lysate and incubated in the dark at
room temperature for 45 min. GSH production was determined at 412
nm using a spectrophotometer (Shimadzu-1640; Shimadzu Corp., Kyoto,
Japan).
Monomer / polymer ratio of
cytoskeletal proteins
PC12 cells were washed with phosphate-buffered
saline (PBS) (3×106 cells/ml) and lysis treated a lysis
buffer containing 50 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol,
phosphatase inhibitors (100 mM Na3VO4 and 10 mM NaF) and a protease
inhibitor (1 mM PMSF). The cell homogenate was incubated with KCl
at a final concentration of 500 mM in the presence of 0.5% v/v
Triton X-100 (Sigma-Aldrich, Munich, Germany). After 2 h of
incubation, the supernatant (namely the monomer) and the pellet
(namely the polymer) were separated by centrifugation at 16,000 ×
g for 20 min at 4°C. Subsequently, the same amounts (50
µg) of supernatant and pellet were loaded onto separate
lanes and separated by 10% SDS-PAGE. The samples were then probed
with antibodies against β-tubulin and β-actin. The integrated
densities of the protein bands were obtained using ImageJ software
(version 1.3; National Institute of Mental Health, Bethesda, MD,
USA) to calculate the ratio of monomer to polymer in β-tubulin and
β-actin under KCl conditions.
AG pretreatment
AG is a nucleophilic hydrazine and nontoxic small
molecule, which exerts pharmacological effects indicating that it
has antioxidant properties (16).
The protective effects of AG have been investigated in several
experimental animal models, including oxidative stress-induced lung
and liver injuries (17), and
ischemia/reperfusion injury (18).
AG appears to have useful properties that may block the protein
carbonylation in PC12 cell lines exposed to
H2O2; therefore, AG pretreatment was
conducted prior to H2O2 exposure in the
present study. PC12 cells in the experimental group were treated
with 0.5 mM AG (AB120123; Abcam, Cambridge, MA, USA) for 30 min,
while PC12 cells in the control group were treated with culture
medium only. After 300 µM H2O2 treatment for
48 h, the changes in the carbonylation level were determined using
western blot analysis.
Transfection
Prior to H2O2 treatment, cells
were suspended at 5×105 cells/ml in DMEM supplemented
with 10% fetal bovine serum and transfected with an empty vector or
the β5 plasmid using the Lipofectamine 2000 reagent, according to
the protocol provided by the manufacturer. At 2 days after
transfection, the expression of β5 in the transfected cells was
examined by western blot analysis.
Detection of carbonylation levels of
β-tubulin and β-actin
A pull-down/western blot method was used to
determine the extent of protein carbonylation. Briefly,
biotinylation of protein carbonyls was performed through reaction
of cells with biotin hydrazide (Bioworld Technology, Inc., St.
Louis Park, MN, USA) in the presence of sodium cyanoborohydride
(Santa Cruz Biotechnology, Santa Cruz, CA, USA). An aliquot (200
µg) of these protein homogenates was kept for western blot
analysis, while streptavidin-agarose (Thermo Fisher Scientific
Inc.) was added to the remainder homogenate in order to isolate the
biotinylated proteins. Next, SDS sample buffer was used to elute
the proteins from the avidin agarose beads, followed by western
blot analysis.
Western blot analysis
At 2 days after transfection, PC12 cells were washed
with phosphate-buffered saline (PBS) and treated with a lysis
buffer containing 50 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol,
phosphatase inhibitors (100 mM Na3VO4 and 10
mM NaF) and a protease inhibitor (1 mM PMSF). The lysates were
centrifuged at 11,000 × g for 10 min at 4°C, and the supernatant
was collected. The protein concentrations were then quantitated
using the Lowry protein assay method. An equal amount of sample (50
µg) was subjected to 10% SDS-PAGE and was blotted onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Subsequently, the samples were blocked in PBS-Tween 20 (PBST)
with 5% nonfat dry milk, and the membranes were incubated with
primary antibodies against β-tubulin (1:1,000) and β-actin
(1:5,000) at appropriate dilutions in PBST overnight at 4°C. The
membranes were then washed three times with PBST solution, followed
by incubation with goat anti-mouse secondary antibody (1:3,000) in
PBST. Subsequently, the proteins were probed with horseradish
peroxidase (HRP)-phytohemagglutinin-L and HRP-concanavalin A
lectin. Visualization of the results was performed by fluorography
using an ECL assay system.
Statistical analysis
Densitometric analysis of the western blot results
was performed by the ImageQuant TL control center (GE Healthcare
Life Sciences). Statistical analysis was performed using GraphPad
Prism version 5 (GraphPad Software, Inc., La Jolla, CA, USA).
One-way analysis of variance was used to compare the parameters
measured in the different study groups.
Results
Increased oxidative stress and
cytoskeletal protein carbonylation in PC12 cells treated with
H2O2
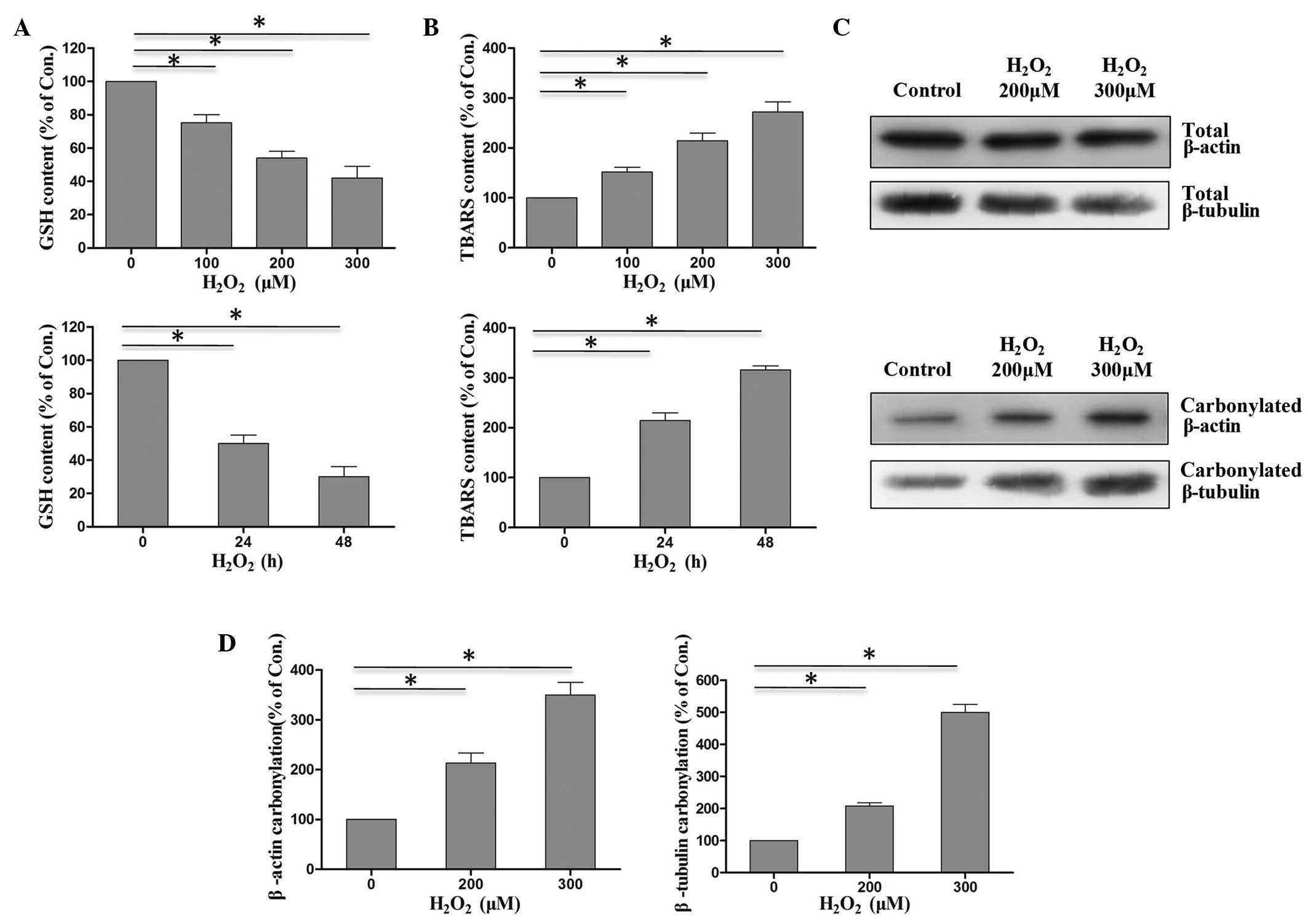
Upon treatment of PC12 cells with increasing
concentrations of H2O2, the level of GSH was
reduced compared with that in the untreated control group (Fig. 1A). The GSH content in PC12 cells was
reduced to 40% compared with the untreated control, and after
treatment with GSH for 24 or 48 h, the GSH content decreased to ~50
and 30%, respectively, which indicated a clear time and
dose-dependent effect. By contrast, the level of TBARS increased
steadily with increasing concentrations of
H2O2, and was ~3-fold higher than the control
group after treatment with 300 µM H2O2 for 48
h (Fig. 1B). These results suggested
that H2O2 induced oxidative stress.
Subsequently, the β-actin and β-tubulin
carbonylation was investigated. A significant increase (P<0.05)
was observed in the proportion of carbonylated β-actin and
β-tubulin (Fig. 1C and D) in
H2O2-treated cells compared with the
untreated control cells, in a dose-dependent manner. Therefore,
these results indicated that the increase in oxidative stress
following TBI may disturb the balance of the reactive carbonyl
species metabolism and lead to carbonylation of cytoskeletal
proteins.
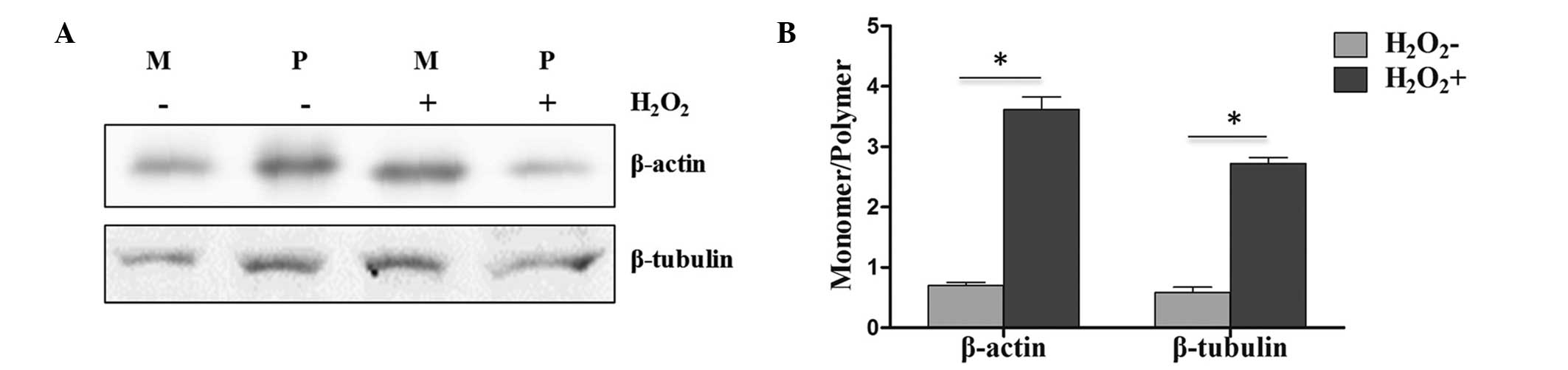
Stability of cytoskeletal proteins in
PC12 cells exposure to H2O2 was affected by
protein carbonylation
Since protein carbonylation is known to affect
cytoskeleton stability, it can be suggested that the observed
changes in cytoskeletal protein carbonylation in PC12 cells treated
with H2O2 have physiological consequences. It
has been reported that carbonylation of tubulin leads to the
disassembly and instability of microtubules, while actin filaments
are easily depolymerized upon carbonylation (19). Thus, in the present study, a series
of assays were performed to examine the functional consequences of
protein carbonylation. As shown in Fig.
2A and B, the results identified that the monomer/polymer
ratios of β-actin and β-tubulin were significantly (P<0.05)
increased with H2O2 treatment, which
suggested dysfunction in the formation of polymers by the
carbonylated β-actin and β-tubulin. These findings indicated that
cytoskeletal proteins could not form stable polymer conditions in
PC12 cells exposed to H2O2, providing
evidence of the functional disturbance in cytoskeletal proteins
under oxidative stress conditions.
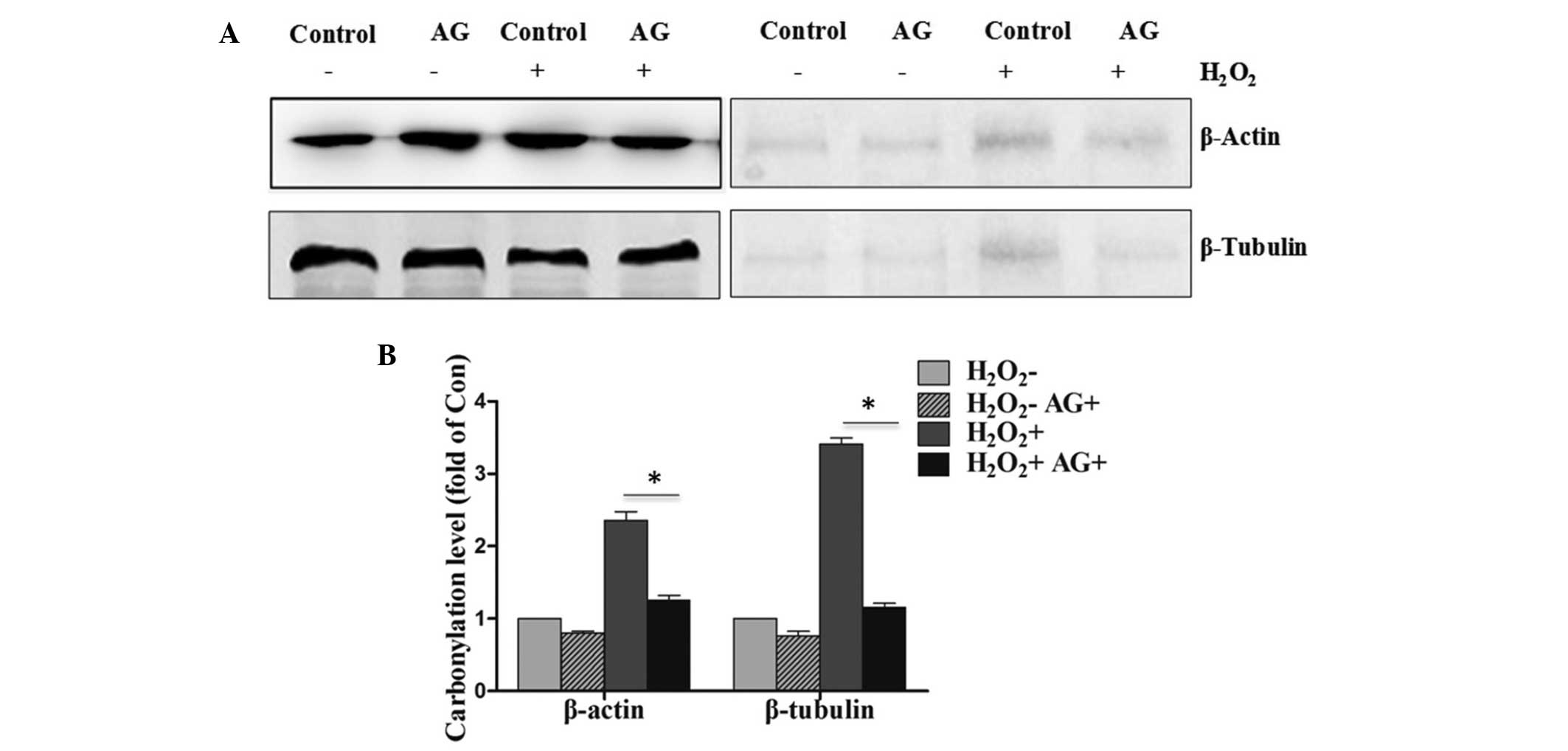
Inhibition of protein carbonylation
upon pretreatment with AG in PC12 cells exposed to
H2O2
The administration of AG at 0.5 h prior to
H2O2 treatment resulted in reduction in the
level of protein carbonylation when compared with that in the
control group (Fig. 3A). In
addition, the pretreatment with AG was found to also block the
proportion of carbonylated β-actin and β-tubulin in PC12 cells
exposed to H2O2. The quantification of the
western blot analysis is shown in Fig.
3B, with a significant difference (P<0.05) observed between
the H2O2-treated and
H2O2+AG-treated groups.
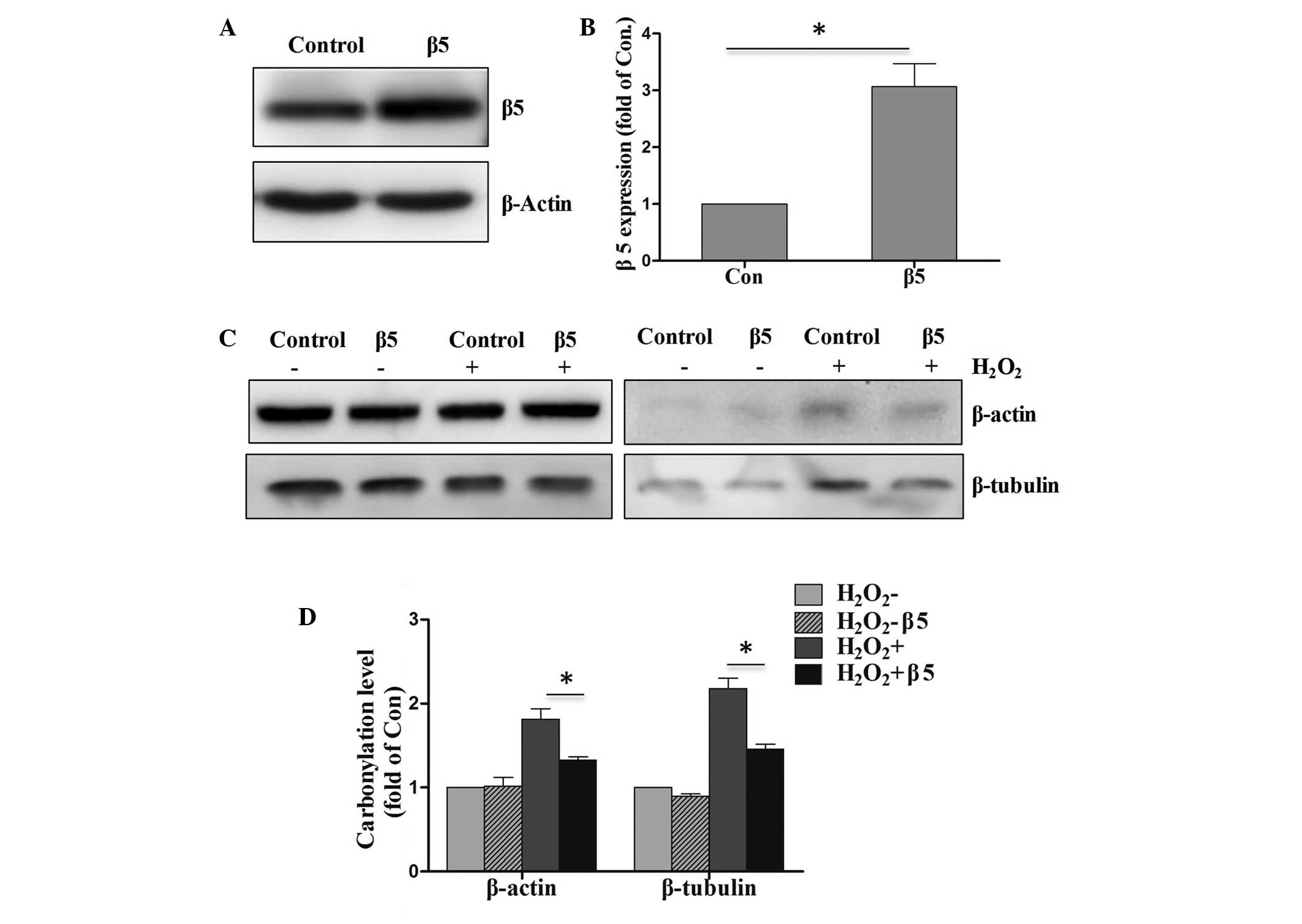
Overexpression of proteasome β5 in
PC12 cells exposed to H2O2 blocks protein
carbonylation
It has been reported that reduced proteasomal
activity contributes to the accumulation of carbonylated proteins
during chronic experimental autoimmune encephalomyelitis in C57BL/6
mice (11). The proteasome serves a
critical role in protein degradation and signal transduction
following cellular stress or tissue injury (20). Overexpression of proteasome β5
assembled subunit increases the amount of proteasome and confers
ameliorated response to oxidative stress and higher survival rates
(21). In order to reveal the effect
of proteasome β5 on the protein carbonylation, plasmids of
proteasome β5 were transfected into PC12 cells. Upregulation of
proteasome β5 in was first observed in the transfected PC12 cells
(Fig. 4A and B). In addition, the
level of protein carbonylation was increased compared with the
control group. The western blot analysis results revealed that the
overexpression of proteasome β5 in PC12 cells also blocked the
proportion of carbonylated β-actin and β-tubulin following cell
exposure to H2O2 (Fig. 4C and D).
Discussion
TBI is a serious health concern often resulting in
mortality, while it may lead to severe neurological dysfunction in
the case of survival (1,2,22). It
has been reported that axonal injury is widely observed in the
development of TBI (7). However,
examining the clinical effectiveness of neuroprotective agents is
challenging.
Free radical-induced oxidative damage reactions, and
in particular membrane lipid peroxidation, are among the best
validated secondary injury mechanisms in preclinical TBI models.
Antioxidants have been demonstrated to alleviate the occurrence of
second injury following TBI (6).
Cytoskeletal proteins, such as GFAP, β-actin and β-tubulin, are
easily carbonylated in cells when exposed to oxidants (14). The present study aimed to identify
the effect of H2O2 exposure, which simulates
the oxidative stress conditions observed in TBI, on the
cytoskeletal proteins in PC12 cells. When the PC12 cells were
cultured with H2O2 in different
concentrations, the increased expression of TBARS and the decreased
expression of GSH indicated increased oxidative stress induced by
the increasing H2O2 concentrations.
Furthermore, the carbonylation levels of β-actin and β-tubulin were
found to be significantly increased in PC12 cells exposed to
H2O2 for 24 and 48 h. In neurodegenerative
disorders, the cytoskeletal proteins are known to be particularly
susceptible to carbonylation (23).
The present study confirmed that cytoskeletal proteins, including
β-actin and β-tubulin, were the target carbonylated proteins when
cells were under oxidative stress. Furthermore, carbonylation of
cytoskeletal proteins has been reported to cause loss of function.
The current study results revealed that, when PC12 cells were
cultured with H2O2, the monomer/polymer
ratios of β-actin and β-tubulin were significantly increased, which
indicated that cytoskeletal β-actin and β-tubulin could not form
stable polymer conditions in PC12 cells exposed to
H2O2, providing evidence of functional
disturbance in cytoskeletal proteins under oxidative stress. Thus,
it can be further postulated that carbonylation will cause
instability of cytoskeletal proteins, thus leading to the axonal
injury in TBI.
In the current study, the carbonylation levels of
β-actin and β-tubulin were blocked following pretreatment with AG
and by overexpression of proteasome β5 in PC12 cells. The principal
function of the proteasome is targeted degradation of intracellular
proteins. The results also revealed that proteasome β5 serves an
important role in the development of TBI. It has been reported that
PA28α overexpression is sufficient to upregulate 11S proteasomes
(24), enhance proteasome-mediated
removal of misfolded and oxidized proteins, as well as protect
against oxidative stress in cardiomyocytes; thus, it can results in
an increase in proteasomal degradation of abnormal cellular
proteins (25). The results of the
present study, thus, indicate the potential value of AG and
proteasome β5 in healing axonal injury in TBI.
In conclusion, the current study suggested that
cytoskeletal proteins carbonylation was involved in the development
of TBI. These findings suggested that the blocking of oxidative
stress-induced carbonylation of cytoskeletal proteins may have a
therapeutic value in the treatment of TBI. However, other factors
may also be associated with the development of TBI, which requires
further investigation.
Acknowledgements
The present study was supported by grants from the
Shenzhen International Cooperation Research Funding (grant no.
GJHZ20120614154914623) and the Shenzhen Key Laboratory of
Neurosurgery (grant no. ZDSYS20140509173142601).
References
|
1
|
Strong K, Mathers C and Bonita R:
Preventing stroke: Saving lives around the world. Lancet Neurol.
6:182–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Langlois JA, Rutland-Brown W and Wald MM:
The epidemiology and impact of traumatic brain injury: A brief
overview. J Head Trauma Rehabil. 21:375–378. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Loane DJ and Faden AI: Neuroprotection for
traumatic brain injury: Translational challenges and emerging
therapeutic strategies. Trends Pharmacol Sci. 31:596–604. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bramlett HM and Dietrich WD: Progressive
damage after brain and spinal cord injury: Pathomechanisms and
treatment strategies. Prog Brain Res. 161:125–141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Petronilho F, Feier G, de Souza B,
Guglielmi C, Constantino LS, Walz R, Quevedo J and Dal-Pizzol F:
Oxidative stress in brain according to traumatic brain injury
intensity. J Surg Res. 164:316–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hall ED, Vaishnav RA and Mustafa AG:
Antioxidant therapies for traumatic brain injury.
Neurotherapeutics. 7:51–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kilinca D, Gallob G and Barbeea KA:
Mechanically-induced membrane poration causes axonal beading and
localized cytoskeletal damage. Exp Neurol. 212:422–430. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Castro JP, Ott C, Jung T, Grune T and
Almeida H: Carbonylation of the cytoskeletal protein actin leads to
aggregate formation. Free Radic Bio Med. 53:916–925. 2012.
View Article : Google Scholar
|
|
9
|
Castro JP, Jung T, Grune T and Almeida H:
Actin carbonylation: From cell dysfunction to organism disorder. J
Proteomics. 92:171–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smerjac SM and Bizzozero OA: Cytoskeletal
protein carbonylation and degradation in experimental autoimmune
encephalomyelitis. J Neurochem. 105:763–772. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng J and Bizzozero OA: Accumulation of
protein carbonyls within cerebellar astrocytes in murine
experimental autoimmune encephalomyelitis. J Neurosci Res.
88:3376–3385. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reed TT, Owen J, Pierce WM, Sebastian A,
Sullivan PG and Butterfield DA: Proteomic identification of
nitrated brain proteins in traumatic brain-injured rats treated
postinjury with gamma-glutamylcysteine ethyl ester: Insights into
the role of elevation of glutathione as a potential therapeutic
strategy for traumatic brain injury. J Neurosci Res. 87:408–417.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dalle-Donne I, Rossi R, Giustarini D,
Gagliano N, Lusini L, Milzani A, Di Simplicia P and Colombo R:
Actin carbonylation: From a simple marker of protein oxidation to
relevant signs of severe functional impairment. Free Radic Biol
Med. 31:1075–1083. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Verma RS, Mehta A and Srivastava N: In
vivo chlorpyrifos induced oxidative stress: Attenuation by
antioxidant vitamins. Pestic Biochem Phys. 88:191–196. 2007.
View Article : Google Scholar
|
|
15
|
Furukawa S, Fujita T, Shimabukuro M, Iwaki
M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M and
Shimomura I: Increased oxidative stress in obesity and its impact
on metabolic syndrome. J Clin Invest. 114:1752–1761. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang F and Iadecola C: Temporal
characteristics of the protective effect of aminoguanidine on
cerebral ischemic damage. Brain Res. 802:104–110. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Majed AA, Khattab M, Raza M,
Al-Shabanah OA and Mostafa AM: Potentiation of diclofenac-induced
anti-inflammatory response by aminoguanidine in carrageenan-induced
acute inflammation in rats: The role of nitric oxide. Inflamm Res.
52:378–382. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alipour M, Gadiri-Soufi F and Jafari MR:
Effect of aminoguanidine on sciatic functional index, oxidative
stress, and rate of apoptosis in an experimental rat model of
ischemia-reperfusion injury. Can J Physiol Pharm. 92:1013–1019.
2014. View Article : Google Scholar
|
|
19
|
Banan A, Fields JZ, Talmage DA, Zhang Y
and Keshavarzian A: PKC-beta1 mediates EGF protection of
microtubules and barrier of intestinal monolayers against oxidants.
Am J Physiol Gastrointest Liver Physiol. 281:G833–G847.
2001.PubMed/NCBI
|
|
20
|
Yao X, Liu J and McCabe JT: Alterations of
cerebral cortex and hippocampal proteasome subunit expression and
function in a traumatic brain injury rat model. J Neurochem.
104:353–363. 2008.PubMed/NCBI
|
|
21
|
Chondrogianni N, Tzavelas C, Pemberton AJ,
Nezis IP, Rivett AJ and Gonos ES: Overexpression of proteasome
beta5 assembled subunit increases the amount of proteasome and
confers ameliorated response to oxidative stress and higher
survival rates. J Biol Chem. 280:11840–11850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schouten JW: Neuroprotection in traumatic
brain injury: A complex struggle against the biology of nature.
Curr Opin Crit Care. 13:134–142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Muntane G, Dalfó E, Martinez A, Rey MJ,
Avila J, Pérez M, Portero M, Pamplona R, Ayala V and Ferrer I:
Glial fibrillary acidic protein is a major target of glycoxidative
and lipoxidative damage in Pick's disease. J Neurochem. 99:177–185.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Powell SR and Wang X: Enhancement of
proteasome function by PA28-alpha; overexpression protects against
oxidative stress. FASEB J. 25:883–893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Neely MD, Boutte A, Milatovic D and
Montine TJ: Mechanisms of 4-hydroxynonenal-induced neuronal
microtubule dysfunction. Brain Res. 1037:90–98. 2005. View Article : Google Scholar : PubMed/NCBI
|