Introduction
Acute lung injury (ALI), characterized by severe
alveolar damage, results from an acute inflammatory response that
leads to edema, neutrophil and macrophage infiltration (1). Development of ALI is a common cause for
admission to critical care units, with incidence in intensive care
units recently reported to be 10.4% (2). Severe sepsis is the most common risk
factor for the development of ALI (2,3).
Hyperoxia therapy is a useful part of treatment for patients with
acute and chronic cardiovascular and pulmonary diseases (4). However, prolonged exposure to hyperoxia
may cause additional deterioration in cases of ALI (5). Acute exposure to hyperoxia induces lung
inflammation and injury, leading to impairment in respiratory
function (6). Prolonged exposure to
high concentrations of oxygen (>50%) may lead to acute or
chronic lung damage, which is characterized by dysfunction of
alveolar epithelial cells (AEC), repressed proliferation and
increased apoptosis and cell death (7,8).
Hyperoxic injury is mediated by accumulation of inflammatory
factors and direct insult resulting from reactive oxygen species
(ROS) (9).
Calcitonin gene-related peptide (CGRP) is a 37 amino
acid neuropeptide that is mainly synthesized and distributed in the
C fibers of sensory nerves in humans and mammals. It is also the
predominant neuromediator to induce vasodilation and neurogenic
inflammation (10). However, a
previous study indicated that CGRP also serves a role in
anti-inflammatory actions and tissue repair, as it decreases
interleukin (IL)-8 secretion, suppresses the formation of ROS and
induces proliferation in the epithelium (11). ROS are often associated with DNA
damage and repair in acute lung injury (12,13).
Previous studies reported that a number of drugs reduce the death
of lung epithelial cells by blocking DNA damage (14–17).
Whether CGRP has the same effects on hyperoxia-injured AEC II is
unknown. The present study was designed to evaluate the role of
CGRP in a hyperoxic cell model (60% oxygen for 24 h) in AEC II
isolated from fetal rats at 19–20 days old, and to investigate
whether the mechanism involved DNA damage repair.
Materials and methods
Suppliers of reagents
Reagents were obtained as follows: Trypsin, DNase I,
Dulbecco's minimal essential medium/F12 (DMEM/F12) and fetal calf
serum (FCS) were obtained from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA); bovine serum albumin (BSA), propidium
iodide and collagenase I was purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany); CGRP and CGRP8–37 were
sourced from Anaspec Inc. (Freemont, CA, USA);
methylenedioxyamphetamine (MDA) and lactate dehydrogenase (LDH)
kits were obtained from Jiancheng Biological Engineering Research
Institute (Nanjing, China); Annexin V-fluorescein isothiocyanate
(FITC) kits were from Jingmei Biotech Co. Ltd. (Shenzhen, China);
TRlzol was from Tiangen Biotech Co., Ltd. (Beijing, China); goat
anti-rabbit immunoglobulin (Ig) G/FITC (cat. no. sc-2012) and
rabbit anti-surfactant protein (SP)-C polyclonal antibody (cat. no.
sc-13979) were from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA); Alexa Fluor 488-conjugated anti-rabbit IgG antibody (cat. no.
A-11008) and Hoechst 33342 were obtained from Molecular Probes
(Thermo Fisher Scientific, Inc.); and rabbit polyclonal γH2AX
(phospho S139) antibody (cat. no. ab2893) was from Abcam
(Cambridge, UK).
Isolation and culture of fetal rat AEC
II
Fetal rat AEC II at the canalicular stage (at 19–20
days of gestation), were isolated using a previously described
method (17). All experiments were
approved by the Animal Care Committee of Kunming Medical
University. Briefly, 30 pregnant Sprague-Dawley rats (Vital River
Laboratories Co., Ltd., Beijing, China), with a mean weight of 264
g (range, 214–307 g) at 19–20 days of gestational were anesthetized
by intraperitoneal injection of pentobarbital (200 mg/kg), and
fetal rats were extracted following onset of adequate anesthesia.
Fetal lungs were lysed and digested with 0.125% trypsin and 10
mg/ml DNase for 25 min at 37°C. Trypsinization was stopped with
DMEM/F12 with 10% FCS and cells were centrifuged at 800 × g for 5
min. Supernatants were removed and cell pellets were resuspended
and incubated in collagenase for 15 min at 37°C. The collagenase
reaction was stopped by adding serum, followed by centrifugation,
as above. Cell pellets were resuspended and plated into 6-well
plates, which were incubated for 1 h at 37°C, for differential
adherence to remove fibroblasts, as non-adherent cells were gently
panned and recovered. Purified cells were plated in 6-well plates
at a seeding density of 1×106 and grown to 70–80%
confluence over 15–18 h in DMEM/F12 supplemented with 10% FBS.
Cultures were maintained at 37°C in a humidified atmosphere
supplemented with 5% CO2 for 24 h, prior to analysis.
The purity of AEC II cells was demonstrated to be >90% by
immunostaining for SP-C (18).
Exposure to air or hyperoxia
AEC II cells were inoculated on cover slides in a
6-well plate and grown to 70–80% confluence. Following this, 10 µM
CGRP or both CGRP and 100 µM CGRP8–37 (a specific CGRP
receptor antagonist), in accordance with pre-test results by
sequential titration, were added into medium prior to exposure to
hyperoxia or air. Hyperoxia was achieved by placing plates in a
modular chamber and filling the chamber with a gas mixture of 60%
oxygen and 5% CO2 until this condition was stable with
the chamber. The ‘air’ condition was accomplished by filling the
chamber with air containing 5% CO2. Following this, the
hyperoxia and air chambers were sealed and put into a 37°C
incubator for 24 h, with continuous monitoring on oxygen fraction
of air using a Pigeon I oxygen measuring meter (Pigeon Medical
Apparatus Co., Ltd., Guangzhou, China).
The experiments were performed in six groups as
follows: i) Air group, cells were cultured in the ‘air’ conditions
(as above); ii) CGRP/air group, cell medium had 10 µM CGRP added 30
min before culturing in air (as above); iii)
CGRP8–37/air group, CGRP and 100 µM CGRP8–37
were added before culturing in air (as above); iv) hyperoxia group,
cells were cultured in the ‘hyperoxia’ conditions (as above); v)
CGRP/O2 group, 10 µM CGRP was added into the medium 30
min before hyperoxia exposure; and vi)
CGRP8–37/O2 group, CGRP and
CGRP8–37 were added into the culture medium 30 min
before hyperoxia treatment.
Immunofluorescence assay for SP-C
AEC II cells were cultured for 15–18 h and treated
with CGRP and CGRP8–37 before exposure to air or 60%
oxygen, as described above. After 24 h, the slides were removed and
rinsed three times with ice-cold PBS. Cells were then fixed with
methanol for 15 min at −20°C, rehydrated twice with PBS, then
blocked with 1% BSA for 10 min at room temperature. After
incubation overnight at 4°C with specific SP-C (1:500) and γH2AX
(phospho S139; 1:200) antibodies, the slides were rinsed
extensively with PBS, and incubated with a FITC-conjugated
secondary antibody (1:1,000) for 1 h at 25°C in the dark.
Visualization was performed using a fluorescence microscope and the
images were analyzed by Image Pro-Plus 5.1 software (Media
Cybernetics, Inc., Rockville, MD, USA). Hoechst 33342 was used as a
nuclear counterstain for automated cell identification, and to
observe nuclear morphology. This counterstain was also used to
determine nuclear size and nuclear staining intensity. Plates were
imaged using the Thermo Scientific ArrayScan XTI HCS Reader (Thermo
Fisher Scientific, Inc.) and analyzed using the Compartmental
Analysis BioApplication (Cellomics, Inc., Thermo Fisher Scientific,
Inc.). Immunostaining-based parameters and nuclear staining-based
parameters were determined and analyzed in the same image set for
each field. For each data replicate, >3 fields of view (a total
of 500 cells) were analyzed.
ROS measurement
Cells were stained with 5 µM cell-permeant
2′,7′-dichlorofluorescin diacetate, an oxidative stress indicator
(Invitrogen; Thermo Fisher Scientific, Inc.), for 15 min at 37°C,
washed in PBS, trypsinized and resuspended at 1×106
cells/ml. The dichlorofluorescein signal was observed and analyzed
with a FACSCalibur flow cytometer and CELLQuest software version
3.3 (BD Biosciences, Franklin Lakes, NJ, USA). Cells were
pre-treated with 10 µM CGRP for 24 h, and incubated with air or 60%
oxygen to examine its effects on ROS production. Cells were
maintained at 37°C for 24 h and dichlorofluorescein was examined in
a 5% oxygen tissue culture incubator to determine the effect of
reduced ambient oxygen on ROS levels.
Apoptosis assay and cell cycle
analysis
AEC II were fixed with 70% ethanol and stored at 4°C
overnight. Prior to staining, the cell suspension was centrifuged
at 500 × g for 5 min, the pellet was washed with PBS and cells were
incubated with propidium iodide for 30 min at 4°C in the dark.
Cellular apoptosis and cell death were detected by Annexin V and PI
staining with an Annexin V-FITC/PI apoptosis detection kit, as
described by the manufacturer. Analysis was performed by flow
cytometry and Flowjo software 7.6 (FlowJo LLC, Ashland, OR, USA)
was used for acquisition and analysis.
Western blotting
AEC II were incubated with RIPA lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China) for 15 min on
ice, then centrifuged at 13,000 × g for 5 min at 4°C. A total of 20
µg protein from cell lysate was separated by 12% SDS-PAGE. Wet gel
system was used to transfer protein samples to a PVDF membrane.
Following 5% slim milk blocking at room temperature for 1.5 h, the
membrane was incubated with a primary anti-caspase 3 antibody (cat.
no. sc-7148; 1:500 dilution; Santa Cruz Biotechnology, Inc.,) at
4°C overnight and subsequently incubated with a secondary antibody
conjugated to horseradish peroxidase (cat. no. sc-2004; 1:2,000
dilution; Santa Cruz Biotechnology, Inc.). β-actin (cat. no.
sc-47778; Santa Cruz Biotechnology, Inc.,) was used as an internal
control. Protein images were observed with an
electrochemiluminescence solution (Pierce; Thermo Fisher
Scientific, Inc.). The experiment was repeated three times.
DNA damage detection by examination of
γH2AX immunofluorescence
ACEII cells were seeded at 2×105
cells/well in black 96-well plates with clear, flat bottoms
(Costar; Corning Incorporated, Corning, NY, USA). Following
treatment, the cells were rinsed with PBS, fixed with 4%
formaldehyde in PBS for 15 min at room temperature, and
permeabilized with 0.1% Triton X-100 in PBS for 10 min. Nonspecific
binding was blocked by incubating the cells with 1% BSA and 0.02%
Triton X-100 in PBS for 20 min at room temperature. The cells were
sequentially incubated with anti-γH2AX antibody (dilution, 1:500)
for 3 h at room temperature, Alexa Fluor 488-conjugated anti-rabbit
IgG antibody (1:500) at room temperature for 1 h, and Hoechst 33342
(10 µg/ml) for 10 min. The cells were washed 3 times with 0.02%
Triton X-100 in PBS for 10 min each time, and were visualized using
an ImageXpress Micro Confocal High-Content Imaging System
(Molecular Devices, LLC, Sunnyvale, CA, USA). Acquisition and
analysis of images, including the number and the total area of
γH2AX foci, were measured using the MetaXpress 4.0.0.24 software
(Molecular Devices, LLC). Images of stained cells were acquired
from the automated fluorescence microscope platform of the
ImageXpress using a 40x objective lens.
Statistical analysis
SPSS 19.0 software (IBM SPSS, Armonk, NY, USA) was
used for statistical analyses. All data are expressed as the mean ±
standard deviation. Group means were compared by analysis of
variance with Tukey's tests used for post hoc analyses.
P<0.05 was considered to indicate a statistically significant
difference.
Results
CGRP reverses the changes in AEC II
that are induced by 60% oxygen
SP-C, secreted only by AEC II, is a biomarker of
these cells (19). Therefore, AEC II
were first plated on slides and the SP-C expression was detected
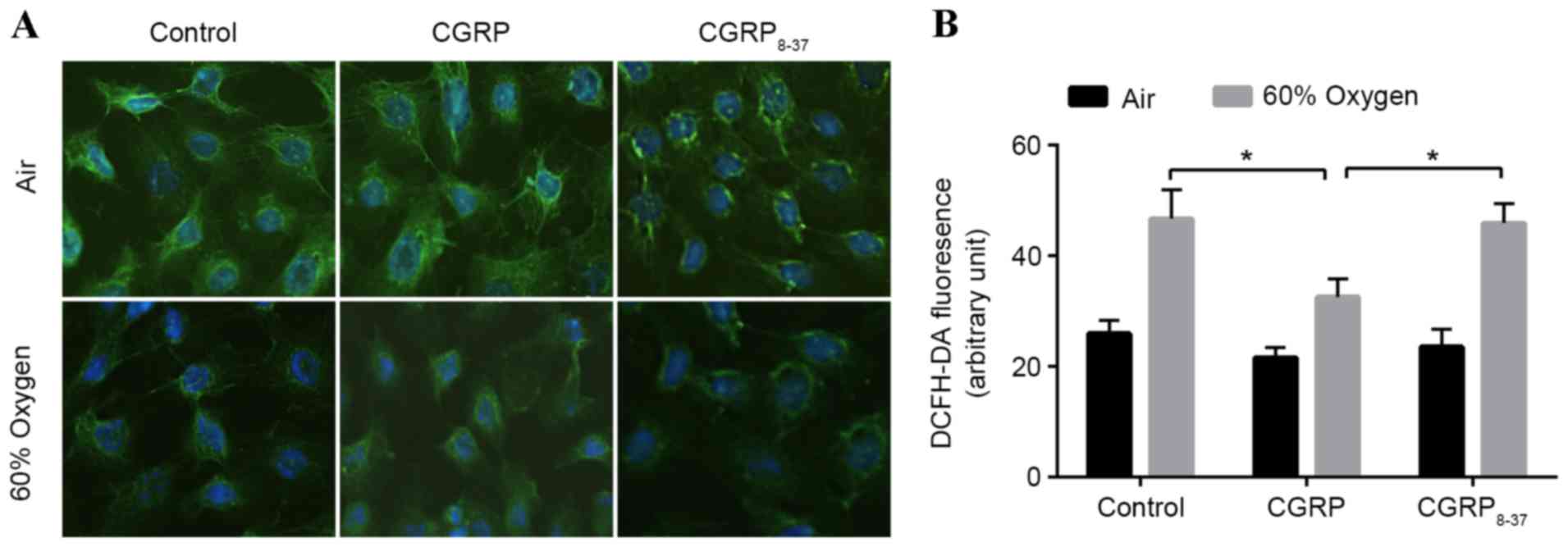
in situ using immunofluorescence (Fig. 1A). SP-C fluorescence in the cytoplasm
was markedly decreased in the hyperoxia groups compared with the
air group, and treatment with CGRP partially rescued
hyperoxia-treated cells, though treatment with CGRP8–37
(the CGRP receptor antagonist) did not. The fluorescence intensity
did not differ among the three groups cultured in air (Fig. 1A). Following each treatment, SP-C
fluorescence in O2 groups was markedly lower than that
in each respective air group.
In order to evaluate the effects of 60% oxygen to
AEC II cells, the levels of ROS in AEC II treated with 60% oxygen
and/or CGRP were assessed using 2′,7′-dichlorofluorescein diacetate
(Fig. 1B). ROS levels were
significantly increased following exposure to 60% oxygen for 24 h
(Fig. 1B). Administration of 10 µM
CGRP prior to hyperoxia significantly inhibited the increase in ROS
levels observed in the hyperoxia group (P<0.05; Fig. 1B). To verify these effects, both CGRP
and CGRP8–37 were applied to competitively block the
binding sites, demonstrating that ROS levels return hyperoxia group
levels. These data indicated a protective effect of CGRP against
hyperoxia insult (Fig. 1B).
CGRP inhibits apoptosis of AEC II that
is induced by hyperoxia
A previous study revealed that 95% oxygen could
induce AEC II apoptosis (20). To
investigate the influence of moderate oxygen on AEC II apoptosis,
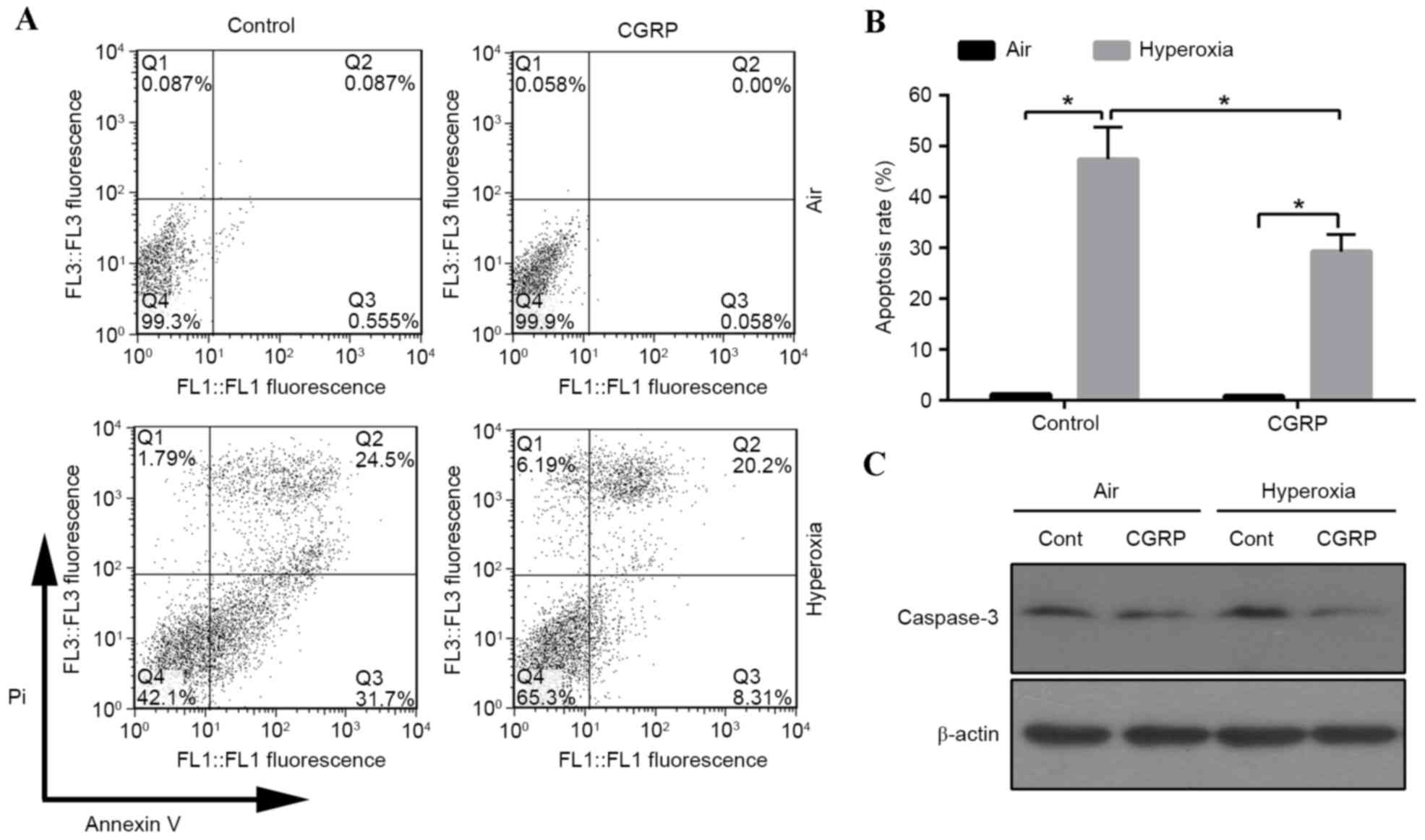
the proportion of apoptotic cells was detected by flow cytometry.
The apoptotic cell number increased in the 60% oxygen groups
compared with the air control groups (Fig. 2A and B). The role of CGRP in
apoptosis was investigated, revealing that the apoptotic rate
significantly decreased in the CGRP/O2 group compared
with the control hyperoxia group (P<0.01; Fig. 2B). Cells treated with high oxygen
expressed active caspase-3, which was inhibited by CGRP treatment
(Fig. 2C). These findings indicated
that hyperoxia triggered apoptosis, which could be inhibited by
CGRP.
Analyses of the relationship between
double strand breaks (DSB) and apoptosis
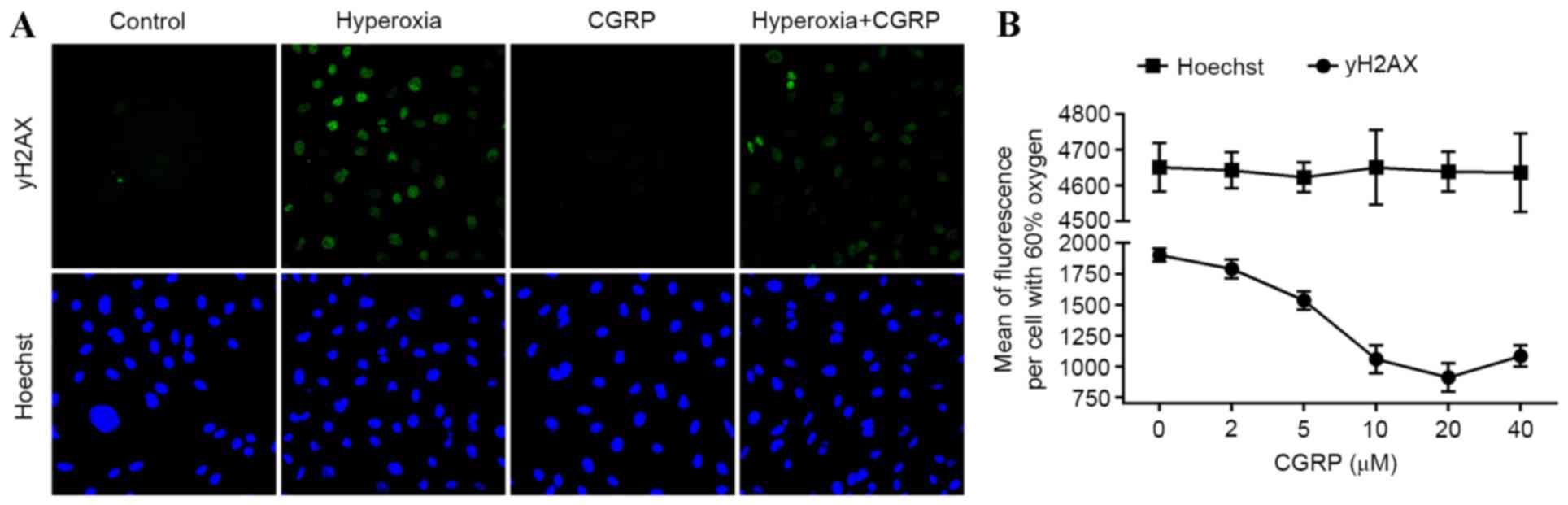
As DSB damage induces apoptosis, cellular senescence
and pro-inflammatory cytokine production (21), the presence of γH2AX, a marker of DSB
and the effects of CGRP were investigated in AEC II. Analysis of
AEC II revealed that the hyperoxia group cells contained higher
numbers of γH2AX foci and CGRP decreased numbers of γH2AX foci
(Fig. 3A). Furthermore, the mean
γH2AX fluorescence measured in these cells demonstrated a
dose-dependent trend following treatment with different
concentrations of CGRP, indicating that DNA fragmentation was
occurring during apoptosis (Fig.
3B). These data suggest that DNA damage is directly linked to
the induction of apoptosis in AEC II.
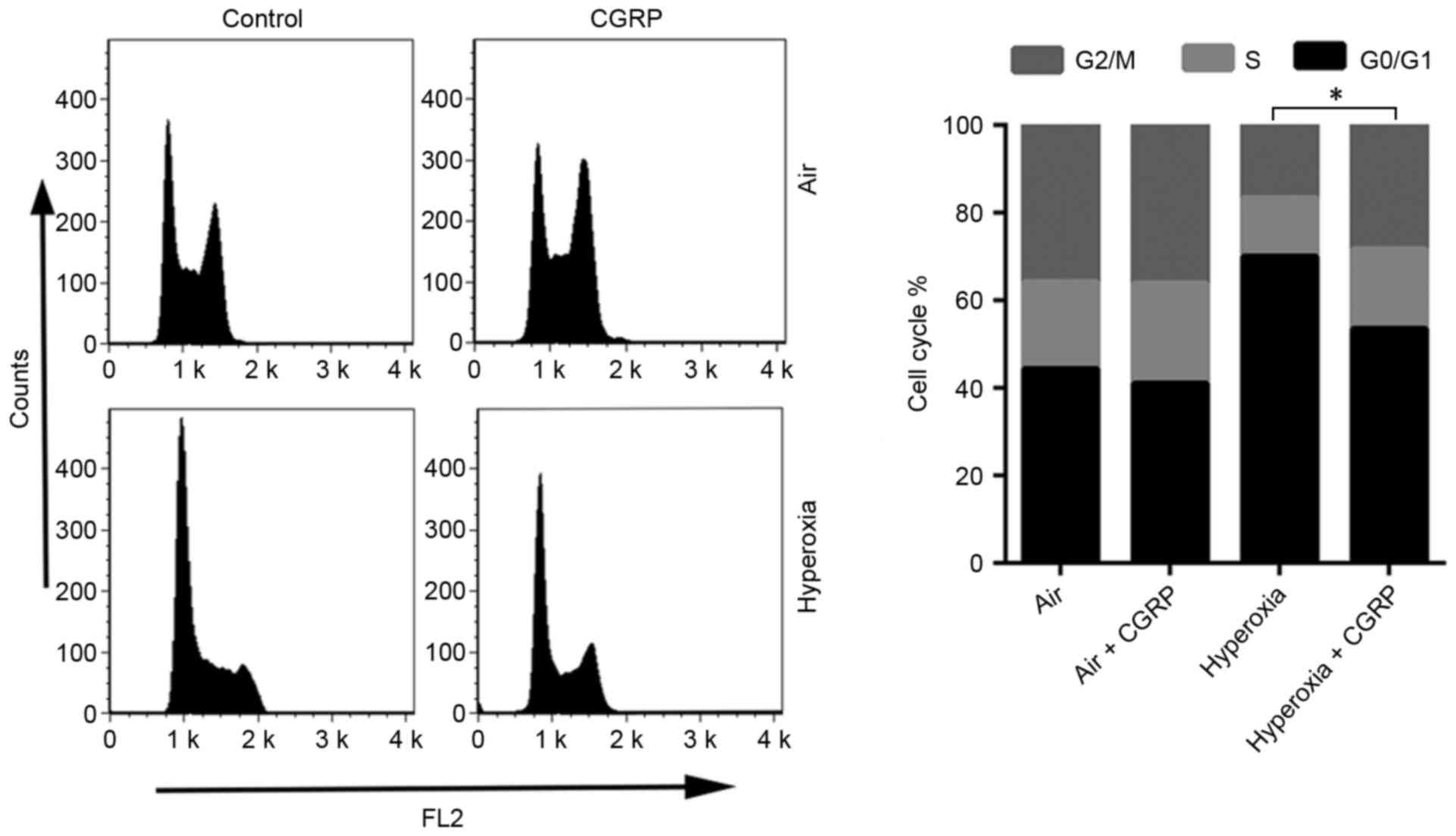
CGRP increases the number of AEC II in
S and G2/M phases
Alterations to the cell cycle often involve DNA
damage and repair (22). For this
reason, the effects of CGRP on the cell cycle of cultured AEC II
were determined. AEC II treated with CGRP (10 µM), and the
proportion of S and G2/M phase cells were detected by flow
cytometry to judge the effects on proliferation (Fig. 4). CGRP at a concentration of 10 µM
significantly enhanced the proportion of cells in the S- and
G2/M-phases, with a consequent reduction of AEC II in G0/G1
(Fig. 4). No significant differences
were observed between the levels of proliferative cells in the air
control and the air + CGRP group.
Discussion
The present study reported that fetal AEC II
exposure to 60% oxygen for 24 h causes cellular injury by inducing
apoptosis, which may be closely associated with the DNA damage
repair mechanism; CGRP may reduce the apoptotic rate induced by
high oxygen concentration by blocking DNA damage.
The pattern of lung injury is closely correlated
with the oxygen concentration and exposure time (23). The model of hyperoxic lung injury,
achieved with an oxygen concentration >85%, lasted for over 2
days (24). Lee et al
(25) isolated AEC II from the lungs
of 95% oxygen-exposed animals and revealed that a sub-lethal
exposure time for this cell line was 48 h, whereas 96 h of exposure
was invariably lethal. Similarly, Pace et al (26) demonstrated that 80% oxygen exposure
for 60 h increased lipid peroxide levels and inflammatory cells in
bronchoalveolar lavage. However, most of those studies were
conducted on AEC II in vivo or as part of a whole lung
homogenate, rather than on cells isolated as a pure population
(5,27,28). In
the present study, purified AEC II cells were exposed to 60% oxygen
for 24 h, and the detrimental effects of moderate oxygen were
investigated. As 60% oxygen is more frequently used than 90% in
clinical practice, 60% oxygen was used to attain a valid hyperoxic
cell model in the current study.
CGRP, mainly expressed in nerve fibers and in the
respiratory system, has multiple effects on cells under
physiological and pathological conditions. It can serve as a
pro-inflammatory factor by stimulating eosinophil migration,
contracting human bronchi and effectively dilating human pulmonary
vessels in vitro (29). In
contrast, other previous reports demonstrated that CGRP suppresses
secretion of inflammatory cytokines, such as IL-6, IL-8 and TNFα,
from macrophages, suggesting its potential anti-inflammatory
properties (30,31). Although CGRP was associated with
regulation of inflammatory cytokines, no previous study has related
this to DNA damage. The present study indicated that CGRP serves a
protective role on hyperoxia-induced AEC II injury by inhibition of
oxidative stress and reduction of apoptosis. H2AX expression was
also detected, which is a biomarker of DNA damage, particularly for
DSB; this revealed increased expression of H2AX in AEC II exposed
into 60% oxygen and decreased H2AX expression in the presence of
CGRP, which could be reversed by addition of the CGRP receptor
antagonist CGRP 8–37.
In addition, to investigate the relationship between
CGRP and DNA damage repair, cell cycle changes were examined. It
was revealed that 10 µM CGRP significantly increased the number of
AEC II in S/G2, both in air and hyperoxia conditions, and that its
antagonist, CGRP 8–37, significantly attenuated the
proliferative effect of CGRP. Previous studies documented that
hyperoxia exerts inhibition of cell growth (32,33)
which is consistent with the current findings that direct exposure
of AEC II to 60% oxygen for 24 h results in an increase of cells in
G0/G1 and a decrease of cells in S and G2/M phase. It is
hypothesized that high oxygen induced DNA damage, leading to AEC II
apoptosis, whilst CGRP reduced DNA damage, enhanced the arrest in S
and G2/M phase, promoted cell repair and inhibited apoptosis.
The exact protective mechanisms of CGRP in AEC II
under hyperoxia exposure have not been identified in the presently
investigated context, and the correlation of CGRP and DNA damage
have not been studied. In the present study, CGRP inhibited cell
death and DNA damage, which may be indirectly affected by other,
associated proteins, rather than direct regulation. A previous
study suggested that CGRP binds to receptors expressed on the cell
surface of AEC II and activates receptor-coupled G proteins,
leading to an induction of intracellular cyclic AMP (cAMP)
generation (34). The accumulated
cAMP inhibits the accumulation of NF-κB complexes in the nucleus by
preventing phosphorylation and degradation of IκB, an NF-κB
inhibitor (35). A previous studies
by the current authors has also demonstrated that high oxygen and
CGRP affects constitutive membrane transport of protein kinase C
(PKC) α, and observed that the activation of NF-κB in the nucleus.
As PKCa and NF-κB appear to serve an important role in apoptosis,
it is hypothesized that CGRP inhibits cell damage and apoptosis by
activating NF-κB or PKCa associated pathways (36). However, a previous study confirmed
that inhibition of NF-κB activity would trigger the protective
mechanism of CGRP to confine the inflammatory response (37). Therefore, it is difficult to identify
whether CGRP inhibited DNA damage via NF-κB, and additional studies
are required to understand the mechanism behind this.
In conclusion, the present study demonstrated that
exposure to 60% oxygen for 24 h predisposed AEC II to oxidative
injury in vitro, including DNA damage and apoptosis;
however, exogenous CGRP markedly attenuated hyperoxic injury and
exerted a cytoprotective effect against hyperoxia insult. This
suggests that upregulation of CGRP expression may represent an
alternative approach for prevention from hyperoxia-induced lung
injury.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30470755 and
81260289). Dr Zijie Liu from The First Affiliated Hospital of
Kunming Medical University (Kunming, Yunnan, China) is thanked for
support in a number of key laboratory techniques.
References
|
1
|
Kallet RH and Matthay MA: Hyperoxic acute
lung injury. Respir Care. 58:123–141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bellani G, Laffey JG, Pham T, Fan E,
Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley
DF, et al: Epidemiology, patterns of care, and mortality for
patients with acute respiratory distress syndrome in intensive care
units in 50 Countries. JAMA. 315:788–800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh G, Gladdy G, Chandy TT and Sen N:
Incidence and outcome of acute lung injury and acute respiratory
distress syndrome in the surgical intensive care unit. Indian J
Crit Care Med. 18:659–665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ward NS, Waxman AB, Homer RJ, Mantell LL,
Einarsson O, Du Y and Elias JA: Interleukin-6-induced protection in
hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 22:535–542.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fukumoto J, Fukumoto I, Parthasarathy PT,
Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen-Gipson DS,
Lockey RF and Kolliputi N: NLRP3 deletion protects from
hyperoxia-induced acute lung injury. Am J Physiol Cell Physio.
305:C182–C189. 2013. View Article : Google Scholar
|
|
6
|
Crapo JD: Morphologic changes in pulmonary
oxygen toxicity. Annu Rev Physiol. 48:721–731. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Durr RA, Dubaybo BA and Thet LA: Repair of
chronic hyperoxic lung injury: Changes in lung ultrastructure and
matrix. Exp Mol Pathol. 47:219–240. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Feinstein SI, Manevich Y, Ho YS
and Fisher AB: Lung injury and mortality with hyperoxia are
increased in peroxiredoxin 6 gene-targeted mice. Free Radic Biol
Med. 37:1736–1743. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warner BB, Stuart LA, Papes RA and Wispe
JR: Functional and pathological effects of prolonged hyperoxia in
neonatal mice. Am J Physiol. 275:L110–L117. 1998.PubMed/NCBI
|
|
10
|
Russo AF: Calcitonin gene-related peptide
(CGRP): A new target for migraine. Annu Rev Pharmacol Toxicol.
55:533–552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li WJ and Wang TK: Calcitonin gene-related
peptide inhibits interleukin-1beta-induced interleukin-8 secretion
in human type II alveolar epithelial cells. Acta Pharmacol Sin.
27:1340–1345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bennett MR: Reactive oxygen species and
death: Oxidative DNA damage in atherosclerosis. Cir Res.
88:648–650. 2001. View Article : Google Scholar
|
|
14
|
Kim SJ, Cheresh P, Williams D, Cheng Y,
Ridge K, Schumacker PT, Weitzman S, Bohr VA and Kamp DW:
Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced
mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem.
289:6165–6176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shao L, Perez RE, Gerthoffer WT, Truog WE
and Xu D: Heat shock protein 27 protects lung epithelial cells from
hyperoxia-induced apoptotic cell death. Pediatr Res. 65:328–333.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Teruya K, Katakura Y, Kabayama S,
Otsubo K, Morisawa S, Ishii Y, Gadek Z and Shirahata S: Effect of
Reduced Water on the Apoptotic Cell Death Triggered by Oxidative
Stress in Pancreatic β HIT-T15 Cell. In: Animal cell technology
meets Genomics. Springer. 2:pp121–124. 2005.
|
|
17
|
Thome UH, Davis IC, Nguyen SV, Shelton BJ
and Matalon S: Modulation of sodium transport in fetal alveolar
epithelial cells by oxygen and corticosterone. Am J Physiol Lung
Cell Mol Physiol. 284:L376–L385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Z, Fang F and Xu F: Effects of
different states of oxidative stress on fetal rat alveolar type II
epithelial cells in vitro and ROSinduced changes in Wnt signaling
pathway expression. Mol Med Rep. 7:1528–1532. 2013.PubMed/NCBI
|
|
19
|
Li F, He J, Wei J, Cho WC and Liu X:
Diversity of epithelial stem cell types in adult lung. Stem Cells
Int. 2015:7283072015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Paepe ME, Mao Q, Chao Y, Powell JL,
Rubin LP and Sharma S: Hyperoxia-induced apoptosis and Fas/FasL
expression in lung epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 289:L647–L659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kidane D, Chae WJ, Czochor J, Eckert KA,
Glazer PM, Bothwell AL and Sweasy JB: Interplay between DNA repair
and inflammation and the link to cancer. Crit Rev Biochem Mol Biol.
49:116–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Branzei D and Foiani M: Regulation of DNA
repair throughout the cell cycle. Nat Rev Mol Cell Biol. 9:297–308.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jankov RP, Luo X, Campbell A, Belcastro R,
Cabacungan J, Johnstone L, Frndova H, Lye SJ and Tanswell AK:
Fibroblast growth factor receptor-1 and neonatal compensatory lung
growth after exposure to 95% oxygen. Am J Respir Crit Care Med.
167:1554–1561. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rogers LK, Tipple TE, Nelin LD and Welty
SE: Differential responses in the lungs of newborn mouse pups
exposed to 85% or >95% oxygen. Pediatr Res. 65:33–38. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee J, Reddy R, Barsky L, Weinberg K and
Driscoll B: Contribution of proliferation and DNA damage repair to
alveolar epithelial type 2 cell recovery from hyperoxia. Am J
Physiol Lung Cell Mol Physiol. 290:L685–L694. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pace PW, Yao LJ, Wilson JX, Possmayer F,
Veldhuizen RA and Lewis JF: The effects of hyperoxia exposure on
lung function and pulmonary surfactant in a rat model of acute lung
injury. Exp Lung Res. 35:380–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Michaelis KA, Agboke F, Liu T, Han K,
Muthu M, Galambos C, Yang G, Dennery PA and Wright CJ:
IκBβ-mediated NF-κB activation confers protection against hyperoxic
lung injury. Am J Respir Cell Mol Biol. 50:429–438. 2014.PubMed/NCBI
|
|
28
|
Vadivel A, Alphonse RS, Ionescu L, Machado
DS, O'Reilly M, Eaton F, Haromy A, Michelakis ED and Thébaud B:
Exogenous hydrogen sulfide (H 2 S) protects alveolar growth in
experimental O2-induced neonatal lung injury. PLoS One.
9:e909652014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Springer J, Geppetti P, Fischer A and
Groneberg DA: Calcitonin gene-related peptide as inflammatory
mediator. Pulm Pharmacol Ther. 16:121–130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nong YH, Titus RG, Ribeiro JM and Remold
HG: Peptides encoded by the calcitonin gene inhibit macrophage
function. J Immunol. 143:45–49. 1989.PubMed/NCBI
|
|
31
|
Li W, Wang T, Ma C, Xiong T, Zhu Y and
Wang X: Calcitonin gene-related peptide inhibits
interleukin-1beta-induced endogenous monocyte chemoattractant
protein-1 secretion in type II alveolar epithelial cells. Am J
Physiol Cell Physiol. 291:C456–C465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yee M, Vitiello PF, Roper JM, Staversky
RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN and
O'Reilly MA: Type II epithelial cells are critical target for
hyperoxia-mediated impairment of postnatal lung development. Am J
Physiol Lung Cell Mol Physiol. 291:L1101–L1111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O'Reilly MA: DNA damage and cell cycle
checkpoints in hyperoxic lung injury: Braking to facilitate repair.
Am J Physiol Lung Cell Mol Physiol. 281:L291–L305. 2001.PubMed/NCBI
|
|
34
|
Drissi H, Lasmoles F, Le Mellay V, Marie
PJ and Lieberherr M: Activation of phospholipase C-beta1 via
Galphaq/11 during calcium mobilization by calcitonin gene-related
peptide. J Biol Chem. 273:20168–20174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brigelius-Flohé R, Banning A, Kny M and
Böl GF: Redox events in interleukin-1 signaling. Arch Biochem
Biophys. 423:66–73. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fu HM, Li L, Wang YJ, Tang CH, Mi HY, Xu F
and Kuang FW: The proliferation-promoting effects of calcitonin
gene-related peptide on type II alveolar epithelial cell exposed to
hyperoxia mediated by protein kinase C alpha pathway. Zhongguo Wei
Zhong Bing Ji Jiu Yi Xue. 22:263–266. 2010.(In Chinese). PubMed/NCBI
|
|
37
|
Piette J, Piret B, Bonizzi G, Schoonbroodt
S, Merville MP, Legrand-Poels S and Bours V: Multiple redox
regulation in NF-kappaB transcription factor activation. Biol Chem.
378:1237–1245. 1997.PubMed/NCBI
|