Introduction
Hemophagocytic lymphohistiocytosis (HLH), also
termed hemophagocytic syndrome, is a state of severe,
life-threatening inflammation caused by an extreme, prolonged and
ineffective immune response (1). HLH
may occur as a genetic or sporadic disorder and, though seen as an
inherited condition affecting primarily a pediatric population, HLH
may occur at any age. Furthermore, HLH may be encountered in
association with a variety of underlying diseases and is considered
a hyperinflammatory syndrome with high mortality, even with
appropriate treatment (1,2). The syndrome is typically induced by
overactive macrophages from the bone marrow or lymph tissue, which
phagocytose erythrocytes, leukocytes and platelets (3,4). The
clinical characteristics of HLH include fever for an extended
duration, hepatosplenomegaly, cytopenia and hemophagocytosis due to
activated macrophages. HLH may be classified as either primary
familial HLH, where the cause is predominantly genetic, or
secondary HLH, which is typically attributed to infections. More
commonly, viral infections such as Epstein-Barr virus (EBV) may
trigger secondary HLH; however, autoimmune diseases and
malignancies have also been demonstrated to have a role in
secondary HLH (5). Previous reports
of HLH have focused on children and malignancy-related diseases
(6,7). HLH in a patient with human
immunodeficiency virus (HIV) infection has rarely been investigated
previously (8). The present study
demonstrates an uncommon case of HLH in combination with HIV
infection in a patient, who eventually succumbed to severe
infection and multiple organ failure following refusal of medical
treatment.
Case report
General information and medical
examination
In May 2015, a 42-year-old male presented to the
First Affiliated Hospital of Nanchang University (Nanchang, China)
with a medical history of high fever experienced for 30 days and
sudden breathing difficulty for 1 day. The family of the patient
complained that 1 month ago, without apparent inducement, he
developed recurrent fever, headache, dizziness, nausea, vomiting,
chest tightness and shortness of breath. The patient was admitted
to a local county-level hospital and given anti-infection and
anti-flu treatment (specific drug use is unknown). The patient
experienced recurrence of high fever for 4–5 days after an initial
improvement in fever symptoms for 2–3 days. This fluctuation in
fever symptoms persisted for several weeks.
Routine blood examination and bone marrow smears
were performed to rule out infectious diseases. The complete blood
count indicated pancytopenia: White blood cell count,
0.30×109/l (normal range, 4–10×109/l); red
blood cell count, 3.22×1012/l (normal range,
4.09–5.71×1012/l); hemoglobin levels, 81 g/l (normal
range, 131–172 g/l); and platelet count, 29×109/l
(normal range, 85–300×109/l). Furthermore, increased
levels of serum ferritin were exhibited (>2,000.0 µg/l; normal
range, 30–400 µg/l) and soluble interleukin-2 receptor levels were
increased (44,000 pg/ml; normal levels, <6,400 pg/ml).
Additional laboratory findings were as follows: White blood cell
count, 0.30×109/l with 20.1% neutrophils, 53.3%
lymphocytes and 23.3% monocytes; aspartate aminotransferase (AST),
721 U/l; alanine aminotransferase (ALT), 130 U/l; lactate
dehydrogenase, 963 mg/dl; alkaline phosphatase, 235 U/l; and
γ-glutamyltranspeptidase, 191 U/l. A reduced level of total protein
was observed (55.2 g/l; normal range, 60–78 g/l), albumin was 21.2
g/l (normal range 34–48 g/l) and there was an increased level of
C-reactive protein (94.60 mg/l; normal range, 0–8 mg/l). D-Dimer
levels were high (6,779 µg/l; normal range, 0–300 µg/l). Serum
antibody to EBV, tubercle bacillus and hemococcidium were
negative.
The results of computed tomography imaging
examination of the upper abdomen indicated infection in both lungs,
fatty liver, splenomegaly and retroperitoneal multiple enlarged
lymph nodes. The findings indicated a preliminary diagnosis of
infectious multiple organ dysfunction syndrome, pulmonary
infection, and hypoproteinemia.
Results of bone marrow and peripheral
blood smear
Bone marrow fluid was obtained by bone marrow
aspiration. A bone marrow smear and peripheral blood smear was
performed using Wright and Giemsa staining (Baso 4017 kit; Baso
Diagnostics Inc., Zhuhai, China). Both samples were observed under
a microscope (ECLIPSE Ci; Nikon Corp., Tokyo, Japan) with a
magnification of ×1,000. Karyocyte cells in the bone marrow were
decreased slightly and the proportion of lymphocytes was high (46%;
normal range, <20%). The proportion of granulocyte and
erythrocytes was borderline normal; however, the mature stage of
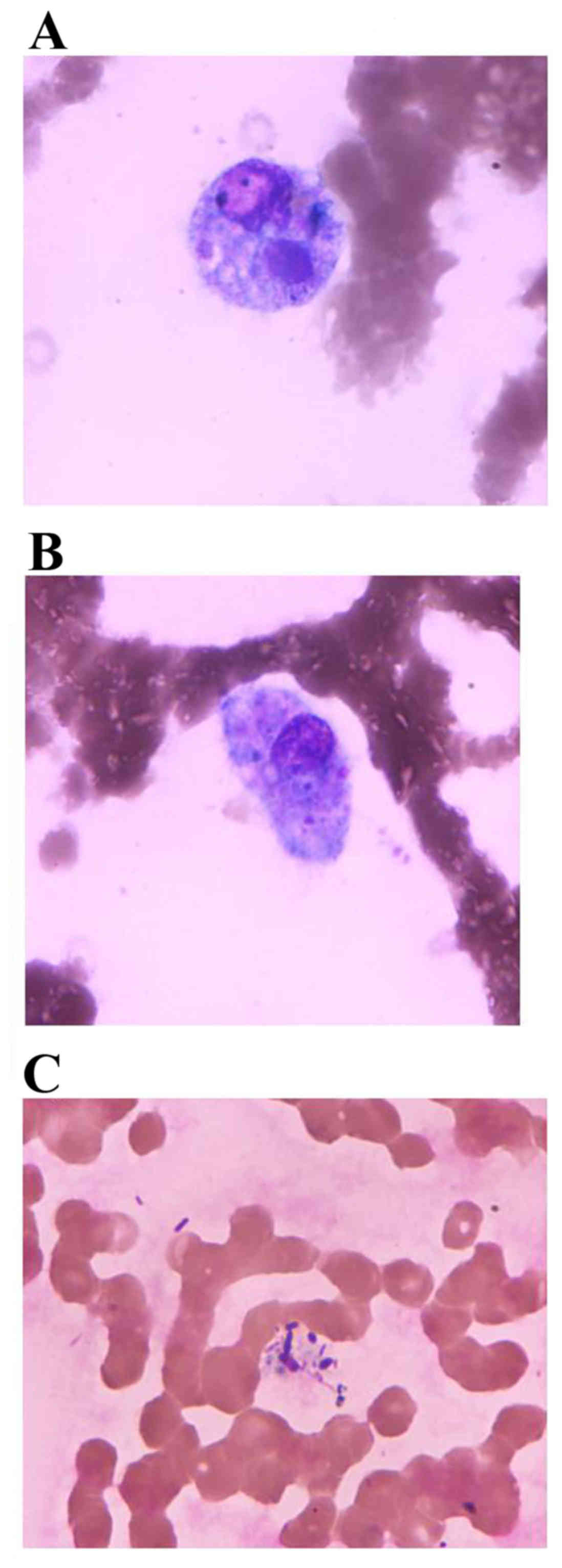
the granulocyte was markedly decreased. Macrophages were exhibited
at an elevated percentage of 8.5% and hemophagocytosis was clearly
observed. Cellular size of the macrophages ranged between 20 and 55
µm in diameter and their shapes were rounded and irregular with
irregular margins accompanying pseudopodia. The macrophage
possessed abundant cytoplasm, indicated as grey blue or light grey,
which engulfed a complete blood cell and fungi or other unknown
microbes (Fig. 1A and B). These
macrophages often exhibited single eccentric nuclei and possessed a
rounded or oval nucleus and loose mesh nuclear chromatin. The
morphology and classification of white blood cells in the
peripheral blood were not abnormal and the distribution of fungi
and bacteria was observed in the cytoplasm (Fig. 1C).
Disease progression and HIV
diagnosis
The patient was hospitalized for three days and
treated with anti-infection and anti-flu treatment (specific agents
unknown). The levels of peripheral blood hemoglobin, percentage of
monocyte cells and platelets decreased and the serum total protein
and albumin levels gradually declined over the course of these
three days. Furthermore, ALT, AST, urea, creatinine and plasma
endotoxin levels were rapidly increased (Table I). On the third day of
hospitalization, the patient appeared delirious, moist rales were
heard in both lungs and multiple organ failure and scattered
ecchymosis were exhibited. The predominant concern was the multiple
organ failure, acute respiratory failure, acute liver failure,
blood coagulation disorder, pulmonary infection and AIDS. Blood
specimens were screened for AIDS and sent for examination to the
Center for Disease Control of Jiangxi Province (CDC) to confirm the
patient was infected with HIV. Following three days, the HIV test
from CDC confirmed a positive diagnosis. The patient's family
refused medical treatment on the third day of hospitalization. The
patient subsequently succumbed to infectious multiple organ failure
in his home.
 | Table I.Routine blood examination results on
each day of hospitalization. |
Table I.
Routine blood examination results on
each day of hospitalization.
| Variable | First day | Second day | Third day |
|---|
| White blood cell
(x109/l) | 0.30 | 0.44 | 0.40 |
| Neutrophils (%) | 20.1 | 20.4 | 17.6 |
| Lymphocytes (%) | 53.3 | 61.4 | 52.5 |
| Monocytes (%) | 23.3 | 18.2 | 16.8 |
| Red blood cells
(x1012/l) | 3.22 | 3.14 | 2.74 |
| Hemoglobin (g/l) | 81 | 78 | 70 |
| Platelets
(x109/l) | 29 | 19 | 13 |
| Alanine
aminotransferase (U/l) | 130 | 147 | 169 |
| Aspartate
aminotransferase (U/l) | 721 | 762 | 943 |
| Total protein
(g/l) | 55.2 | 42.3 | 38.5 |
| Albumin (g/l) | 21.2 | 20.5 | 19.5 |
| Globulin (g/l) | 34.0 | 21.8 | 19.0 |
| Ratio of albumin to
globulin | 0.62 | 0.94 | 1.03 |
|
γ-glutamyltranspeptidase (U/l) | 191 | 156 | 122 |
| Creatinine
(mmol/l) | 77.7 | 293.0 | 285.7 |
| Urea (µmol/l) | 4.2 | 7.5 | 10.3 |
| Uric acid
(µmol/l) | 118 | 255 | 248 |
| Creatine kinase
(U/l) | – | 199 | 573 |
| Amylase (U/l) | – | 116 | 123 |
| Plasma endotoxin
(U/ml) | 1.23 | 3.06 | 23.58 |
Discussion
Fever, cytopenia of at least two cell types,
hypertriglyceridemia and/or hypofibrinogenemia, hyperferritinemia
(>500 µg/l), hemophagocytosis, elevated levels of serum CD25,
decreased levels of NK cell activity and splenomegaly are a set of
symptoms, of which five are required to suggest secondary HLH,
according to the guidelines of the International Histiocyte Society
(9). Based on the clinical and
laboratory findings of fever, splenomegaly, cytopenias,
hemophagocytosis in the bone marrow, hyperferritinemia and raised
serum CD25 levels, a diagnosis of secondary HLH was therefore
established.
The association between HLH and HIV is not well
understood, as few such cases exist. HIV infection may contribute
to the development of HLH, possibly through mechanisms related to
CD4+ cells (predominantly T lymphocytes, monocytes,
macrophages and dendritic cells) (10). Following HIV infection, viral
incorporation of CD4+ cell DNA, combined with an
overwhelming trophic response of relapsed T cell lymphoma may
overstimulate the immune system, leading to HLH (11). At present, the pathogenesis of HLH
has been suggested to be associated with a deficiency in cytolytic
activity, which results from stimulation of lymphocytes and
histiocytes (12). This uncontrolled
immune response causes enhanced production of pro-inflammatory
cytokines and major histocompatibility complex I and II molecules
from macrophages as well as the expansion of inflammatory
monocytes. Subsequently, this heightened inflammatory response
causes necrosis, organ failure and promotes the proliferation and
phagocytic activity of histiocytes (13). In the present case, the macrophages
appeared to exhibit hyperactivity, which was indicated by the
markedly elevated percentage of monocytes detected in the
peripheral blood and the markedly increased percentage of
macrophages detected in the bone marrow. Furthermore,
hemophagocytosis in macrophages was clearly observed from the bone
marrow and peripheral blood smear.
HLH is difficult to treat and is typically
associated with a high morbidity and mortality rate (3,14). HLH
therapy must target the suppression the hyper-stimulated immune
system via abolishing activated CD8+ T lymphocytes or
macrophages, in addition to treating any existing HLH triggers,
including infections, autoimmune diseases and malignancies
(14).
The present case study demonstrates that HLH
associated with HIV infection is a severe disease with a poor
prognosis and may result in infection, multiple organ failure and
mortality if the correct treatment is not administered in a
timely-manner. In the present case, laboratory testing, including
cell morphological examination and hemophagocytosis detected in the
bone marrow, indicated that HLH was associated with HIV infection.
Therefore, laboratory physicians should consider HLH and identify
the possible cause (infections, autoimmune disease and
malignancies) once intense hemophagocytosis is detected in the bone
marrow.
References
|
1
|
Janka GE and Lehmberg K: Hemophagocytic
lymphohistiocytosis: Pathogenesis and treatment. Hematology Am Soc
Hematol Educ Program. 2013:605–611. 2013.PubMed/NCBI
|
|
2
|
Campo M and Berliner N: Hemophagocytic
Lymphohistiocytosis in adults. Hematol Oncol Clin North Am.
29:915–925. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Janka GE and Lehmberg K: Hemophagocytic
syndromes-an update. Blood Rev. 28:135–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malinowska I, Machaczka M, Popko K,
Siwicka A, Salamonowicz M and Nasiłowska-Adamska B: Hemophagocytic
syndrome in children and adults. Arch Immunol Ther Exp (Warsz).
62:385–394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lehmberg K, Albert MH, Beier R, Beutel K,
Gruhn B, Kröger N, Meisel R, Schulz A, Stachel D, Woessmann W, et
al: Treosulfan-based conditioning regimen for children and
adolescents with hemophagocytic lymphohistiocytosis. Haematologica.
99:180–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lehmberg K, Nichols KE, Henter JI,
Girschikofsky M, Greenwood T, Jordan M, Kumar A, Minkov M, La Rosée
P, Weitzman S, et al: Consensus recommendations for the diagnosis
and management of hemophagocytic lymphohistiocytosis associated
with malignancies. Haematologica. 100:997–1004. 2015.PubMed/NCBI
|
|
8
|
Adachi E, Koibuchi T, Imai K, Kikuchi T,
Shimizu S, Koga M, Nakamura H, Iwamoto A and Fujii T:
Hemophagocytic syndrome in an acute human immunodeficiency virus
infection. Intern Med. 52:629–632. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meki A, O'Connor D, Roberts C and Murray
J: Hemophagocytic lymphohistiocytosis in chronic lymphocytic
leukemia. J Clin Oncol. 29:e685–e687. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Usman M, Thapa SD, Hadid H and Yessayan
LT: HIV infection presenting proliferation of CD8+ T lymphocyte and
hemophagocytic lymphohistiocytosis. Int J STD AIDS. 27:411–413.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uemura M, Huynh R, Kuo A, Antelo F, Deiss
R and Yeh J: Hemophagocytic Lymphohistiocytosis complicating T-cell
lymphoma in a patient with HIV infection. Case Rep Hematol.
2013:6872602013.PubMed/NCBI
|
|
12
|
Tothova Z and Berliner N: Hemophagocytic
syndrome and critical Illness: New insights into diagnosis and
management. J Intensive Care Med. 30:401–412. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rouphael NG, Talati NJ, Vaughan C,
Cunningham K, Moreira R and Gould C: Infections associated with
haemophagocytic syndrome. Lancet Infect Dis. 7:814–822. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Janka G: Hemophagocytic
lymphohistiocytosis: When the immune system runs amok. Klin
Padiatr. 221:278–285. 2009. View Article : Google Scholar : PubMed/NCBI
|