Introduction
Childhood epilepsy is a common neurological
condition and affects ~0.5–1% in the general population, with an
approximate total of 50 million people worldwide (1). Early-onset epileptic encephalopathies
are considered to be severe neurological disorders, which lead to
impaired motor, cognitive, and sensory development due to
recurrence of seizures. It has been recognized that many of the
observed epilepsy phenotypes are associated with specific
chromosomal imbalances and thus, display gene dosage effects, which
are of significant interest to understand the specific roles of the
involved genes and their cognate protein products (2). Therefore, information on the
chromosomal abnormalities related to epilepsy likely reveal the
underlying mechanisms for various epilepsy phenotypes, including
fragile-X, trisomy 12p, Wolf-Hirschhorn, ring 20, and 1p36 deletion
syndromes. Even though the role of genetic factors in idiopathic
epilepsies has been suggested for a long time, the involvement of
genetic factors has been clearly demonstrated in cryptogenic and
symptomatic epilepsies (3). Nearly
40% of the etiological causes for epilepsy are now known to be due
to genetic factors (4). However,
Mendelian epilepsies seem to account only for 1% of epilepsies,
implicating non-Mendelian inheritance of some of the affected
genes. Interestingly, the estimated risk of epilepsy for
off-springs and siblings of epileptic patients was found to be only
2–5% (3). In fact, advanced
next-generation high throughput sequencing technologies, have led
to the recognition of several new candidate genes that may play a
role in the pathogenesis of early-onset epileptic encephalopathies
(5,6).
The role of environmental factors in genetic
epilepsies cannot be discounted, even though the major determining
factor for the seizures is the underlying mutation, since the
environmental factors can influence the expression of such genetic
defect, phenotypically either aggravating it, or even dampening it
(7). On the basis of genetic
complexity, there appear to be two types of epilepsies: i) The
Mendelian epilepsies, which are generally monogenic, simple, and
rare and account for ~1% of all epilepsy cases (8) and ii) complex epilepsies, which affect
the majority of the patients, are common and multigenic. Several
studies have suggested that genetic and environmental factors
interact to different degrees and thereby influence the
susceptibility to epilepsy (9).
Mendelian epilepsies include monogenic syndromes with single gene
defects causing the specific phenotype (9) and often show variable penetrance and
severity and these also include the majority of cases of Dravet
syndrome [severe myoclonic epilepsy of infancy (SMEI)], caused by
de novo mutations (10). The
monogenic inheritance of epilepsy syndromes include autosomal,
X-linked, and mitochondrial types, although most common epilepsies
display complex or polygenic inheritance (11). Some epilepsy syndromes, such as
epileptic encephalopathies, involve intractable seizures in
association with intellectual decline, and include Lennox-Gastaut
syndrome, myoclonic astatic epilepsy of Doose, and Dravet syndrome
(12). Epileptic encephalopathies
can be due to acquired etiologies or structural abnormalities of
the involved proteins like ion channels or they may be because of
specific gene mutations affecting neuronal excitability. Increased
understanding of the molecular and genetic insights of these
syndromes hold promise for designing disease-specific
therapies.
Chromosome 18 abnormalities in epilepsy
Nearly 500 different chromosomal abnormalities have
been described to be associated with disturbed EEG patterns and
seizures. Certain chromosomal abnormalities show a greater
association with epilepsy, e.g., Wolf-Hirschhorn (4p-),
Miller-Dieker (del 17p13.3), Angelman syndrome (del 15q11-q13),
inversion duplication 15, and ring chromosomes 14 and 20, while
many others show weaker associations (13). Rearrangements of chromosome 18 have
been observed in some patients; however, due to the lack of
complete details on the epilepsy it is difficult to draw precise
genotype-phenotype correlations. Nevertheless, patients with
trisomy or duplication of chromosome 18 were found to display high
incidence of epileptic seizures with a prevalence of up to 65%
(14). Children with 18p-deletion,
show an anomaly as they have less frequent epileptic seizures but
poor seizure control. On the other hand, patients with 18q-deletion
syndrome also suffer from epilepsy with predominantly focal
seizures, which are well controllable and occurring during early
years of life. Among all the abnormalities of chromosome 18,
18q-deletion syndrome and full trisomy 18 are described to be
frequently associated with epilepsy. Patients with trisomy 18 show
both partial and generalized epilepsies, with onset in the first
year of life (13).
Channelopathies
Several ion channels are essential for proper
maintenance of neuronal excitability and these include
voltage-gated sodium, potassium, and chloride ion channels and
ligand-gated acetylcholine receptor and γ-aminobutyric acid
subunit-α receptor-mediated ion channels. Mutations in the genes
coding for these ion-channel components lead to ion channel
dysfunction, also known as channelopathies, which are the basis of
the development of several epilepsy syndromes (15). The mutations causing channelopathies
can be either a gain or a loss of ion channel function and
contribute to the pathogenesis of epilepsy syndrome (16,17).
Benign familial neonatal seizures (BNFS) have been found to be
associated with mutations in the genes KCNQ2 and KCNQ3 that code
for M-channel subunits of voltage-gated potassium channels, present
in the central nervous system (18).
A mutation in either the α4-subunit (CHRNA4) or the β2-subunit
(CHRNB2) gene of the neuronal nicotinic acetylcholine receptor are
known to cause the partial seizures associated with autosomal
dominant nocturnal frontal lobe epilepsy (19). More common are the mutations that
affect the function of sodium channels such as voltage-gated sodium
channel, type 1 (Nav 1.1) and lead to the epilepsy
syndromes in children (20).
Sodium channel mutations
Nav channels, which exist in three
different functional states (open, closed and inactivated states)
are responsible for the initiation and propagation of neuronal
action potentials (21). At rest, Na
channels are in closed conformation and no sodium current flows.
Upon membrane depolarization, the channel opens with a rapid influx
of Na+ ions into the neuron. The Na+ ion
current peaks in <1 msec and falls back in few milliseconds.
Soon after opening, the channels are closed by a gating mechanism
to prevent continuous membrane depolarization. There are 9 subtypes
of Na channels (Nav 1.1 1.1-Nav 1.9), and
Nav 1.1, which is encoded by the SCN1A gene, is densely
distributed in the initial segments of axons in the brain, the site
of generation of action potentials (22,23). Na
channels regulate the action potential output of the neuron, which
also determines the excitability of neighboring neurons. Thus, Na
channels influence not only the overall excitability of neural
networks but also of the individual neighboring neurons. Therefore,
an imbalance in the excitation and inhibition in the Na channel
operated neuronal excitation can lead to uncontrolled neuronal
firing, hyperexcitability, and seizures. Linkage analysis of an
Australian pedigree spanning 4 generations, comprising generalized
epilepsy with febrile seizures plus (GEFS+) syndrome (24) led to identification of a mutation in
SCN1B, on chromosome 19, which codes for the β1-subunit of the
Nav1.1 channel (25). This mutation,
which causes a tryptophan for cysteine substitution disrupts the
formation of a putative disulphide bridge in the extracellular
immunoglobulin G loop of the β-subunit, thus causing accelerated Na
current flow through the Na channel and thus membrane
hyperexcitability (26).
Subsequent studies identified several mutations in
SCN1A and SCN1B genes of Na channels causing channelopathies
(27) in patients with GEFS+
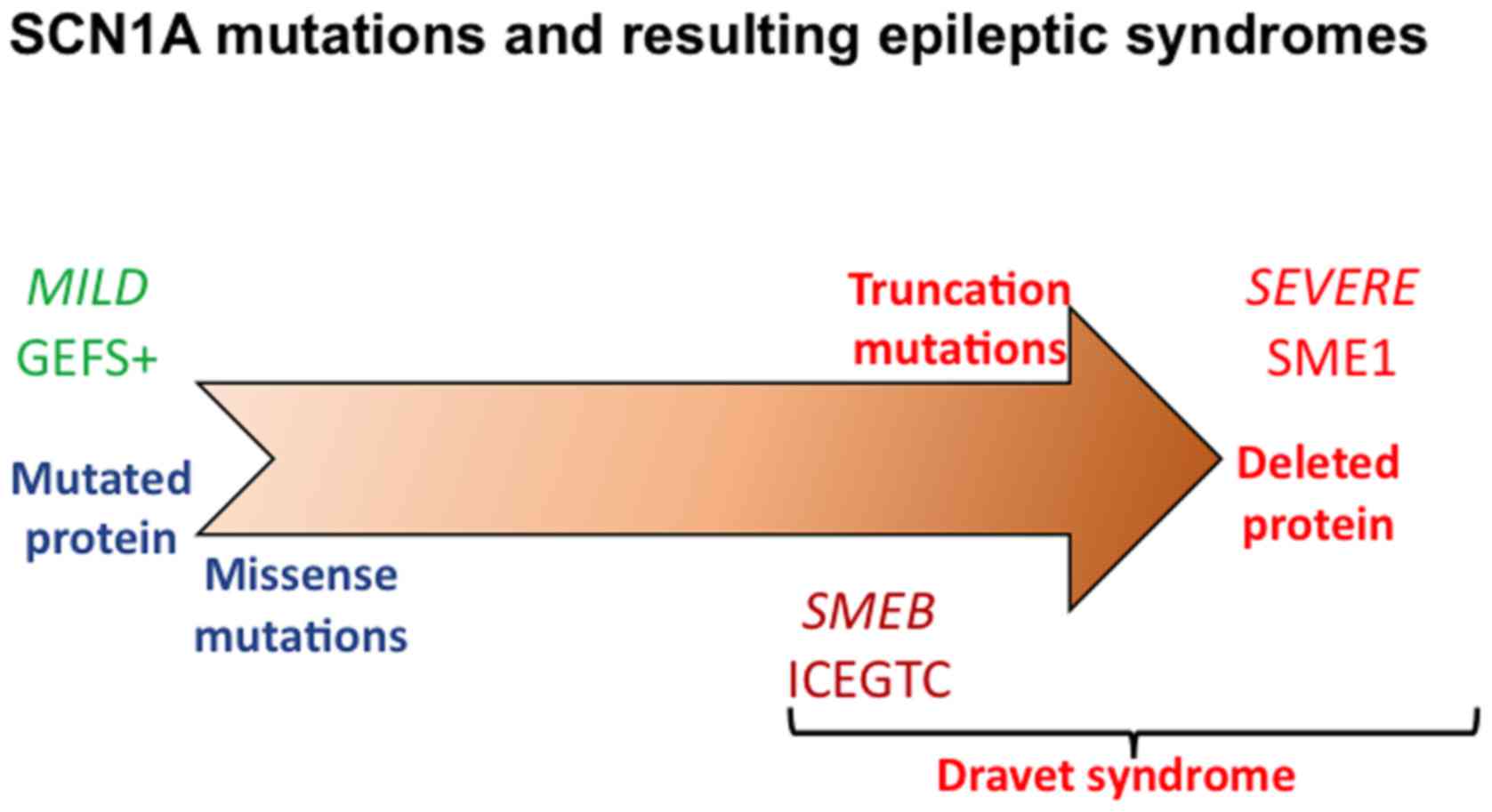
(Fig. 1). Apparently, only ~10% of
the families with GEFS+ display mutations in the SCN1A gene, which
are missense mutations, with single amino acid substitutions.
Besides GEFS+, mutations in SCN1A have also been observed in almost
80% of the patients with SMEI, where these mutations occur de
novo (28). In patients with
SMEI, 40–67% of SCN1A mutations are nonsense or frame-shift types
that cause truncation of the coded protein. Missense mutations of
SCN1A gene are ~30–40%, whereas ~20% mutations are deletions and
splice site mutations in SMEI (28).
Thus, SCN1A mutations can lead to a broad spectrum of epileptic
syndromes (Fig. 1) ranging from mild
(GEFS+) to severe (SMEI), with the intermediary borderland clinical
syndromes, depending upon the type of mutation, with truncation
mutations being most severe and missense mutations that cause
single amino acid substitutions being less severe (29). It is evident that patients with Na
channelopathies should not be administered with Na channel blockers
for any other indication, as these drugs can worsen their
situation. At present, genetic testing for these mutations is
highly expensive and not readily accessible, even though it is
desirable to be available for routine clinical testing of the
affected patients.
Pathogenic heterozygous mutations in another gene,
SCN8A, which codes for a 1980 amino acid Na channel protein (Nav1.6
subunit) that is involved in membrane depolarization during the
generation of action potentials in neurons and also in muscles
(30), are also found to cause both
early onset epileptic seizures and intellectual disability without
epilepsy. It has been found through whole exome sequencing that
majority of the SCN8A mutations are de novo and display a
wide clinical pattern that includes multiple seizure types,
intellectual disability as well as developmental retardation
following the onset of seizures (31).
BFNS
BFNS are clusters of seizures, which appear within
the first few days after birth up to the third month and disappear
spontaneously, with a 15% risk of seizures appearing later in life
(32). Most patients suffering with
this form of epilepsy syndrome have mutated KCNQ2, the gene
encoding the voltage-dependent K+ channel, KQT-like subtype member,
and deletions/duplications involving one or more exons of this gene
(32). Mutations in the associated
gene KCNQ3, voltage-dependent K+ channel, KQT-like subtype member 3
are seen in relatively fewer families (17). In vitro studies revealed that
heteromeric wild-type and mutant KCNQ2⁄3 channels, when coexpressed
show a ~30% reduction in the potassium current, which accounts for
the development of BFNS (33).
However, it is not apparent why these seizures are seen only in
neonates and it is speculated that this may be due to higher
susceptibility of neurons in neonatal brain and/or because of
possible replacement of the mutated channels in the later stages of
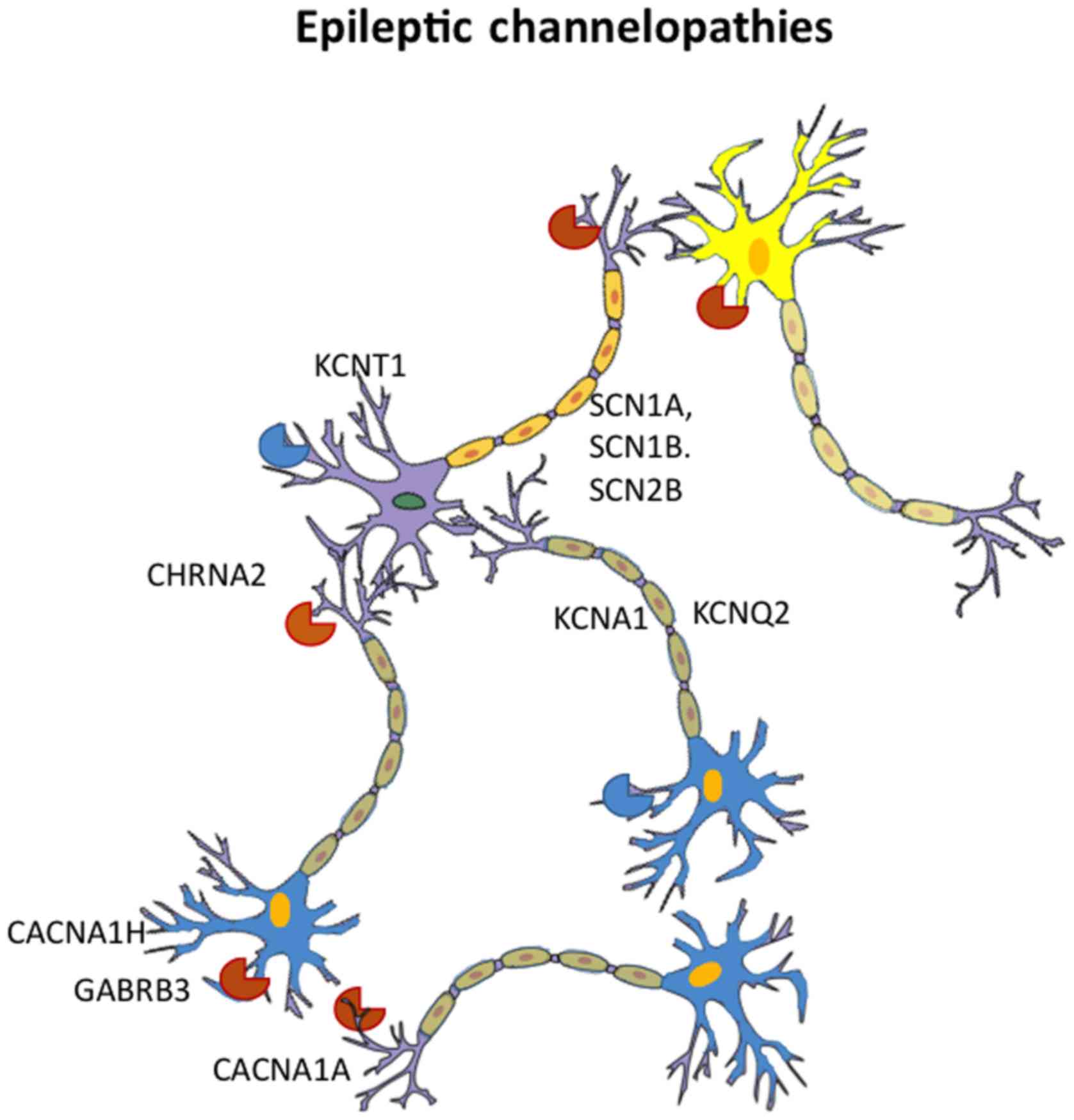
growth (34). Mutations in other ion
channel protein coding genes including KCNB1 gene that encodes the
KV2.1 potassium channel (35), KCNT1
gene that encodes a sodium-activated potassium channel (36), CACNA2D2 gene, encoding for an
auxiliary subunit of high voltage-gated calcium channels, which is
involved in the regulation of the protein trafficking and,
subsequently, of neuronal calcium current influx (37), and the HCN1 gene that encodes a
hyperpolarization-activated cation channel (38), have been found to be associated with
different forms of epilepsy (Fig.
2).
Benign familial infantile seizures are similar to
those observed in BNFS, but have an age of onset ~6 months
(39), and these patients display
mutations of the PRRT2 gene, at the pericentromeric region of
chromosome 16, and also suffer from familial infantile convulsions,
paroxysmal kinesigenic or exercise-induced dyskinesia, migraine, or
hemiplegic migraine, or in various combinations (40,41).
However, the location or the type of mutation of PRRT2 gene do not
appear to affect the severity of the disease (41). PRRT2 codes for a protein that
interacts with a synaptosomal membrane protein involved in
Ca2+ triggered exocytosis.
Non-ion channel genes and epileptic
encephalopathies
Even though the majority of epileptic syndromes have
been found to be associated with mutated ion channel proteins, few
non-ion channel proteins are also found to be involved in epileptic
syndromes. PCDH19, which is part of the protocadherin delta-2
subclass of the cadherin super family and expressed mostly in the
nervous system, where it plays a role in neuronal connections, is
found to be mutated in an X-linked epilepsy and mental disorder
that affects only girls (42). Other
cases of non-ion channel genes that are implicated in epileptic
syndromes include: Aristaless-related homeobox gene, on chromosome
Xp22, a transcription factor that belongs to a family of paired
class homeobox genes and involved in the nervous system development
(43); X-linked gene
cyclin-dependent, kinase-like 5, mutations of which cause severe
myoclonic form of epilepsy (44);
and syntaxin binding protein 1 gene, mutation of which is
associated with Ohtahara syndrome or early infantile eplileptic
encephalopathy (45).
Recent whole exome sequencing studies revealed many
other genes that are found mutated in different forms of epileptic
encephalopathies. Most of these mutations have been thought to
arise as de novo mutations or are inherited in an autosomal
recessive fashion, as compound heterozygous mutations (31). Mutations of these new genes, which
are associated with different forms of epilepsy phenotypes, are
found in a limited number of cases and sometimes resemble the known
syndromes, including Dravet syndrome (46,47).
Management of epilepsy syndromes and
therapeutic approaches
There are several significant co-morbid features
associated with epileptic encephalopathy and these include the loss
of language or other cognitive or developmental abilities,
behavioral and attention deficits including autistic-like features,
psychiatric problems, and sleep disorders (48). Management of epilepsy is often
challenging, and requires treatment of not only the seizures but
also the other frequently associated disabling co-morbidities.
Unfortunately, the conventional anti-epileptic medications are far
from effective in most patients. However, few drugs appear to be
promising in specific cases. Thus, vigabatrin is found to be quite
effective in West syndrome, if caused by tuberous sclerosis. But
this drug has a major drawback as it poses the risk of vigabatrin
associated visual loss (49). Among
other medications, valproate and lamotrigine are considered as a
first-line treatment in Lennox-Gastaut syndrome (LGS) (50). Also rufinamide use in a randomized
controlled trial in LGS patients led to a marked reduction of
seizures (51). Clobazam is
frequently used to treat Dravet syndrome and LGS patients.
Benzodiazepine therapy may have a risk of worsening tonic seizures
in LGS patients. Sulthiame, a carbonic anhydrase inhibitor, which
acts via Na channels has also been used in some forms of epilepsies
(52). Besides these drugs,
corticosteroids have been used in childhood epilepsies but only a
small proportion of patients respond to this therapy (53,54).
Interestingly, ketogenic diet, which consists of high ratio of fat
to carbohydrate, has been found to be efficacious in reducing
multiple forms of epilepsies and it has been suggested that use of
this diet early in infancy may be more effective (55,56).
However, the mechanism by which ketogenic diet offers this
protection from seizures is not known yet. In patients who are
resistant to pharmacological interventions, surgery is often
considered as a palliative option. Patients with focal or
hemispheric lesions might improve in response to surgical
treatment, such as lobectomy, hemispherotomy, or hemispherectomy,
particularly in earlier stage (57,58).
Advances in cell biology will likely help in future in the use of
induced pluripotent stem cells, to correct the genetic defects that
lead to the various forms of epileptic syndromes. However, this
approach is still in the lab and may take some time to reach the
clinic.
Conclusions
Early-onset epileptic encephalopathies are severe
neurological disorders, which lead to impaired motor, cognitive,
and sensory development. Several chromosomal abnormalities and gene
mutations, particularly those coding for ion channel proteins have
been implicated in the pathogenesis of epileptic encephalopathies.
High throughput sequencing technologies and whole exome sequencing
have led to the recognition of several new candidate genes, besides
those coding for ion channel proteins, with a role in the
pathogenesis of epileptic encephalopathies. A significant quantum
of study has been done on the role of SCN1A Na channel coding gene
in the pathogenesis of different forms of epilepsy. Despite the
identification of several players, an effective therapy for
epilepsy is not yet available and the currently used medications
suffer from unwanted side effects. Corticosteroids and ketogenic
diets have been found to be better in managing epilepsy, when
implemented early in life. Breakthrough in genetic engineering and
gene editing technologies and stem cell applications should
hopefully lead to a better therapy for epilepsy not far from
now.
References
|
1
|
Pal DK, Pong AW and Chung WK: Genetic
evaluation and counseling for epilepsy. Nat Rev Neurol. 6:445–453.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elia M, Musumeci SA, Ferri R and Ayala GF:
Chromosome abnormalities and epilepsy. Epilepsia. 42:(Suppl 1).
24–27; discussion 28. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peljto AL, Barker-Cummings C, Vasoli VM,
Leibson CL, Hauser WA, Buchhalter JR and Ottman R: Familial risk of
epilepsy: A population-based study. Brain. 137:795–805. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guerrini R and Noebels J: How can advances
in epilepsy genetics lead to better treatments and cures? Adv Exp
Med Biol. 813:309–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nieh SE and Sherr EH: Epileptic
encephalopathies: New genes and new pathways. Neurotherapeutics.
11:796–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Helbig I: New technologies in molecular
genetics: The impact on epilepsy research. Prog Brain Res.
213:253–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scheffer IE: Epilepsy genetics
revolutionizes clinical practice. Neuropediatrics. 45:70–74. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tripathi M and Jain S: Genetics in
epilepsy: Transcultural perspectives. Epilepsia. 44:(Suppl 1).
12–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thomas RH and Berkovic SF: The hidden
genetics of epilepsy-a clinically important new paradigm. Nat Rev
Neurol. 10:283–292. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Novarino G, Baek ST and Gleeson JG: The
sacred disease: The puzzling genetics of epileptic disorders.
Neuron. 80:9–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reid CA, Berkovic SF and Petrou S:
Mechanisms of human inherited epilepsies. Prog Neurobiol. 87:41–57.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nabbout R and Dulac O: Epileptic syndromes
in infancy and childhood. Curr Opin Neurol. 21:161–166. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verrotti A, Carelli A, di Genova L and
Striano P: Epilepsy and chromosome 18 abnormalities: A review.
Seizure. 32:78–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heron SE, Cox K, Grinton BE, Zuberi SM,
Kivity S, Afawi Z, Straussberg R, Berkovic SF, Scheffer IE and
Mulley JC: Deletions or duplications in KCNQ2 can cause benign
familial neonatal seizures. J Med Genet. 44:791–796. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Helbig I, Scheffer IE, Mulley JC and
Berkovic SF: Navigating the channels and beyond: Unravelling the
genetics of the epilepsies. Lancet Neurol. 7:231–245. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Steinlein OK: Genetic mechanisms that
underlie epilepsy. Nat Rev Neurosci. 5:400–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Waxman SG: Channel, neuronal and clinical
function in sodium channelopathies: From genotype to phenotype. Nat
Neurosci. 10:405–409. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh NA, Westenskow P, Charlier C, Pappas
C, Leslie J, Dillon J, Anderson VE, Sanguinetti MC, Leppert MF and
Consortium BP: BFNC Physician Consortium: KCNQ2 and KCNQ3 potassium
channel genes in benign familial neonatal convulsions: Expansion of
the functional and mutation spectrum. Brain. 126:2726–2737. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steinlein OK, Mulley JC, Propping P,
Wallace RH, Phillips HA, Sutherland GR, Scheffer IE and Berkovic
SF: A missense mutation in the neuronal nicotinic acetylcholine
receptor alpha 4 subunit is associated with autosomal dominant
nocturnal frontal lobe epilepsy. Nat Genet. 11:201–203. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ragsdale DS: How do mutant Nav1.1 sodium
channels cause epilepsy? Brain Res Brain Res Rev. 58:149–159. 2008.
View Article : Google Scholar
|
|
21
|
George AL Jr: Inherited disorders of
voltage-gated sodium channels. J Clin Invest. 115:1990–1999. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Catterall WA, Dib-Hajj S, Meisler MH and
Pietrobon D: Inherited neuronal ion channelopathies: New windows on
complex neurological diseases. J Neurosci. 28:11768–11777. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Palmer LM and Stuart GJ: Site of action
potential initiation in layer 5 pyramidal neurons. J Neurosci.
26:1854–1863. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scheffer IE and Berkovic SF: Generalized
epilepsy with febrile seizures plus. A genetic disorder with
heterogeneous clinical phenotypes. Brain. 120:479–490. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wallace RH, Wang DW, Singh R, Scheffer IE,
George AL Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland
GR, et al: Febrile seizures and generalized epilepsy associated
with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat
Genet. 19:366–370. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stafstrom CE: Severe epilepsy syndromes of
early childhood: The link between genetics and pathophysiology with
a focus on SCN1A mutations. J Child Neurol. 24:15S–23S. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sugawara T, Tsurubuchi Y, Agarwala KL, Ito
M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada
K, et al: A missense mutation of the Na+ channel alpha II subunit
gene Na(v)1.2 in a patient with febrile and afebrile seizures
causes channel dysfunction. Proc Natl Acad Sci USA. 98:6384–6389.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujiwara T, Sugawara T, Mazaki-Miyazaki E,
Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K,
Yamakawa K, et al: Mutations of sodium channel alpha subunit type 1
(SCN1A) in intractable childhood epilepsies with frequent
generalized tonic-clonic seizures. Brain. 126:531–546. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lossin C: A catalog of SCN1A variants.
Brain Dev. 31:114–130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Veeramah KR, O'Brien JE, Meisler MH, Cheng
X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE,
Restifo LL, et al: De novo pathogenic SCN8A mutation identified by
whole-genome sequencing of a family quartet affected by infantile
epileptic encephalopathy and SUDEP. Am J Hum Genet. 90:502–510.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Larsen J, Carvill GL, Gardella E, Kluger
G, Schmiedel G, Barisic N, Depienne C, Brilstra E, Mang Y, Nielsen
JE, et al: EuroEPINOMICS RES Consortium CRP: The phenotypic
spectrum of SCN8A encephalopathy. Neurology. 84:480–489. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guerrini R, Marini C and Mantegazza M:
Genetic epilepsy syndromes without structural brain abnormalities:
Clinical features and experimental models. Neurotherapeutics.
11:269–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maljevic S, Wuttke TV, Seebohm G and
Lerche H: KV7 channelopathies. Pflugers Arch. 460:277–288. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weber YG and Lerche H: Genetic mechanisms
in idiopathic epilepsies. Dev Med Child Neurol. 50:648–654. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Srivastava S, Cohen JS, Vernon H, Barañano
K, McClellan R, Jamal L, Naidu S and Fatemi A: Clinical whole exome
sequencing in child neurology practice. Ann Neurol. 76:473–483.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barcia G, Fleming MR, Deligniere A, Gazula
VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio
R, et al: De novo gain-of-function KCNT1 channel mutations cause
malignant migrating partial seizures of infancy. Nat Genet.
44:1255–1259. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Edvardson S, Oz S, Abulhijaa FA, Taher FB,
Shaag A, Zenvirt S, Dascal N and Elpeleg O: Early infantile
epileptic encephalopathy associated with a high voltage gated
calcium channelopathy. J Med Genet. 50:118–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nava C, Dalle C, Rastetter A, Striano P,
de Kovel CG, Nabbout R, Cancès C, Ville D, Brilstra EH, Gobbi G, et
al: EuroEPINOMICS RES Consortium: De novo mutations in HCN1 cause
early infantile epileptic encephalopathy. Nat Genet. 46:640–645.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vigevano F: Benign familial infantile
seizures. Brain Dev. 27:172–177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marini C, Conti V, Mei D, Battaglia D,
Lettori D, Losito E, Bruccini G, Tortorella G and Guerrini R: PRRT2
mutations in familial infantile seizures, paroxysmal dyskinesia,
and hemiplegic migraine. Neurology. 79:2109–2114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guerrini R and Mink JW: Paroxysmal
disorders associated with PRRT2 mutations shake up expectations on
ion channel genes. Neurology. 79:2086–2088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dibbens LM, Tarpey PS, Hynes K, Bayly MA,
Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C,
et al: X-linked protocadherin 19 mutations cause female-limited
epilepsy and cognitive impairment. Nat Genet. 40:776–781. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gécz J, Cloosterman D and Partington M:
ARX: A gene for all seasons. Curr Opin Genet Dev. 16:308–316. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Buoni S, Zannolli R, Colamaria V, Macucci
F, di Bartolo RM, Corbini L, Orsi A, Zappella M and Hayek J:
Myoclonic encephalopathy in the CDKL5 gene mutation. Clin
Neurophysiol. 117:223–227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ohtahara S and Yamatogi Y: Ohtahara
syndrome: With special reference to its developmental aspects for
differentiating from early myoclonic encephalopathy. Epilepsy Res.
70:(Suppl 1). S58–S67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Carvill GL, Heavin SB, Yendle SC, McMahon
JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S,
et al: Targeted resequencing in epileptic encephalopathies
identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet.
45:825–830. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Suls A, Jaehn JA, Kecskés A, Weber Y,
Weckhuysen S, Craiu DC, Siekierska A, Djémié T, Afrikanova T,
Gormley P, et al: EuroEPINOMICS RES Consortium: De novo
loss-of-function mutations in CHD2 cause a fever-sensitive
myoclonic epileptic encephalopathy sharing features with Dravet
syndrome. Am J Hum Genet. 93:967–975. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
McTague A and Cross JH: Treatment of
epileptic encephalopathies. CNS Drugs. 27:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maguire MJ, Hemming K, Wild JM, Hutton JL
and Marson AG: Prevalence of visual field loss following exposure
to vigabatrin therapy: A systematic review. Epilepsia.
51:2423–2431. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lemmon ME and Kossoff EH: New treatment
options for lennox-gastaut syndrome. Curr Treat Options Neurol.
15:519–528. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Glauser T, Kluger G, Sachdeo R, Krauss G,
Perdomo C and Arroyo S: Rufinamide for generalized seizures
associated with Lennox-Gastaut syndrome. Neurology. 70:1950–1958.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fejerman N, Caraballo R, Cersósimo R,
Ferraro SM, Galicchio S and Amartino H: Sulthiame add-on therapy in
children with focal epilepsies associated with encephalopathy
related to electrical status epilepticus during slow sleep (ESES).
Epilepsia. 53:1156–1161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Goldsmith IL, Zupanc ML and Buchhalter JR:
Long-term seizure outcome in 74 patients with Lennox-Gastaut
syndrome: Effects of incorporating MRI head imaging in defining the
cryptogenic subgroup. Epilepsia. 41:395–399. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nunes VD, Sawyer L, Neilson J, Sarri G and
Cross JH: Diagnosis and management of the epilepsies in adults and
children: Summary of updated NICE guidance. BMJ. 344:e2812012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li B, Tong L, Jia G and Sun R: Effects of
ketogenic diet on the clinical and electroencephalographic features
of children with drug therapy-resistant epilepsy. Exp Ther Med.
5:611–615. 2013.PubMed/NCBI
|
|
56
|
Thammongkol S, Vears DF, Bicknell-Royle J,
Nation J, Draffin K, Stewart KG, Scheffer IE and Mackay MT:
Efficacy of the ketogenic diet: Which epilepsies respond?
Epilepsia. 53:e55–e59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cross JH and Neville BG: The surgical
treatment of Landau-Kleffner syndrome. Epilepsia. 50:(Suppl 7).
63–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Peltola ME, Liukkonen E, Granström ML,
Paetau R, Kantola-Sorsa E, Valanne L, Falck B, Blomstedt G and
Gaily E: The effect of surgery in encephalopathy with electrical
status epilepticus during sleep. Epilepsia. 52:602–609. 2011.
View Article : Google Scholar : PubMed/NCBI
|