Introduction
Atrial fibrillation (AF), the most common sustained
cardiac arrhythmia in clinical practice, is able to induce cardiac
dysfunction and strokes (1).
Oxidative stress contributes to the genesis of AF (2) and oxidative modifications of proteins
are found in chronic AF patients (3). Reactive oxygen species (ROS) may result
in abnormal Ca2+ handling and changes in mitochondrial
function, leading to arrhythmogenesis (4–8).
Mitochondria are key regulators of cardiomyocyte energy metabolism
and redox state control (8).
Mitochondrial dysfunction-elicited ROS production was proposed as
the basis of the mitochondrial free-radical theory of aging
(9–12). Evidence indicates that mitochondrial
dysfunction may directly alter cardiomyocyte excitability and
cell-to-cell coupling through regulating the adenosine
monophosphate protein kinase, the adenosine triphosphate-sensitive
potassium channel and the sarcolemmal sodium channel (13–16).
Furthermore, coenzyme (Co)-Q10, an agent beneficial for
mitochondrial function, is widely used to treat heart failure and
ischemic heart diseases, which are critical risk factors of AF
(17–19). However, it is not clear whether
Co-Q10 has a role in modifying the effects of
mitochondrial dysfunction in atrial arrhythmogenesis.
Pulmonary veins (PVs), subsidiary pacemakers, which
contain a mixture of working myocardium and pacemaker cells, are an
important source of AF initiation and maintenance (20–22).
Sinoatrial node (SAN) dysfunction may enhance PV arrhythmogenesis,
which may contribute to the high incidence of AF during sick sinus
syndrome (23). A previous study has
demonstrated that the right and left atria (RA and LA) have
different electrical responses to hypoxia and reoxygenation, a
condition that may cause mitochondrial dysfunction (24). Therefore, the aim of the present
study was to investigate whether mitochondrial dysfunction
differentially regulates electrical activity between SANs and PVs
or between the RA and LA.
Materials and methods
Ethics statement
The present investigation was approved by the
Institutional Animal Care and Use Committee of the National Defense
Medical Center (Taipei, Taiwan; IACUC-15-297) and conformed to the
institutional Guide for the Care and Use of Laboratory Animals
published by the U.S. National Institutes of Health.
Rabbit SAN, PV and atrial tissue
preparations
As previously described (2,23), all
of the rabbits had ad libitum access to food and water, were
maintained in a temperature and humidity-controlled environment
(20–22°C; 50–70% humidity) with a 12 h light/dark cycle, and were
raised in stainless steel cages. A total of 20 male New Zealand
rabbits (Animal Health Research Institute, New Taipei City, Taiwan)
weighing 1.5–2.0 kg and aged 3–4 months were anesthetized with an
intravenous injection of sodium pentobarbital (100 mg/kg of body
weight), followed by an intravenous injection of heparin (1,000
IU/kg of body weight). Subsequently, a midline thoracotomy was
performed and the heart and lungs were removed. For dissection of
the PVs, the LA was opened by an incision along the mitral valve
annulus extending from the coronary sinus to the septum in Tyrode's
solution, composed of 137 mM NaCl, 4 mM KCl, 15 mM
NaHCO3, 0.5 mM NaH2 PO4, 0.5 mM
MgCl2, 2.7 mM CaCl2 and 11 mM dextrose. The
PV was separated from the atrium at the level of the LA-PV junction
and separated from the lungs at the ending of the PV myocardial
sleeves. One end of the preparation, consisting of the PV and
atrial-PV junction, was pinned with needles to the bottom of a
tissue bath. The other end (distal PV) was connected to a Grass
FT03C force transducer with a silk thread. The adventitia or
epicardial side of the preparation faced upwards. LA and RA tissues
were prepared from the LA (10.0×5.0×0.5 mm) and RA appendages
(10.0×5.0×0.5 mm), respectively. For SAN-PV tissue preparations,
the SAN with the RA and the right superior PV with the LA were
isolated. Tissue preparations were superfused with normal Tyrode's
solution and were left to equilibrate for 1 h prior to
electrophysiological study.
Electrophysiological and
pharmacological studies
Transmembrane action potentials (APs) of the SAN,
PVs, RA and LA were recorded using machine-pulled glass capillary
microelectrodes filled with 3 M KCl. Preparations were connected to
a WPI model FD223 electrometer under a tension of 150 mg.
Electrical and mechanical events were simultaneously displayed on a
Gould 4072 oscilloscope and Gould TA11 recorder. Signals were
digitally recorded with a 16-bit accuracy at a rate of 125 kHz. An
electrical stimulus with a 10-msec duration and supra-threshold
strength was provided by a Grass S88 stimulator through a Grass
SIU5B stimulus isolation unit.
For the SAN-PV interaction study, transmembrane APs
of the PVs and SANs were recorded within 3 mm of the distal part of
the PV myocardial sleeve and the SAN by simultaneously using
machine-pulled glass capillary microelectrodes filled with 3 M KCl,
which were connected to a WPI model FD223 electrometer. Tissue was
superfused at a constant rate (3 ml/min) with Tyrode's solution
saturated with a 97% O2/3% CO2 gas mixture.
The temperature was maintained at 37°C and the preparations were
left to equilibrate for 1 h prior to initiation of the
electrophysiological study. Electrical events were simultaneously
displayed on a Gould 4072 oscilloscope and a Gould TA11 recorder.
Signals were digitally recorded with a 16-bit accuracy at a rate of
125 kHz. Trifluorocarbonylcyanide phenylhydrazone (FCCP; a
mitochondrial uncoupling agent) at 10, 100 and 300 nM with and
without Co-Q10 (at 10 µM) was perfused for 20 min to
test the pharmacological responses of the PV and SAN in the intact
SAN-PV preparation. Spontaneous activity was defined as the
constant occurrence of spontaneous APs in the absence of any
electrical stimuli.
AP amplitude (APA) was obtained from the resting
membrane potential or maximum diastolic potential to the peak of AP
depolarization. AP durations (APDs) at repolarization of 20, 50 and
90% of the APA were measured as the APD20,
APD50 and APD90, respectively. Spontaneous
activity was defined as the constant occurrence of spontaneous APs
in the absence of any electrical stimuli.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. A repeated one-way analysis of variance with post-hoc
Tukey's test was used to compare the effects of FCCP on the RA and
LA. The effects of FCCP and Co-Q10 on the PV and SAN
were compared by a Wilcoxon signed-rank test or a paired t-test,
depending on the outcome of the normality test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of FCCP on the electrical
activity in isolated PVs and SANs
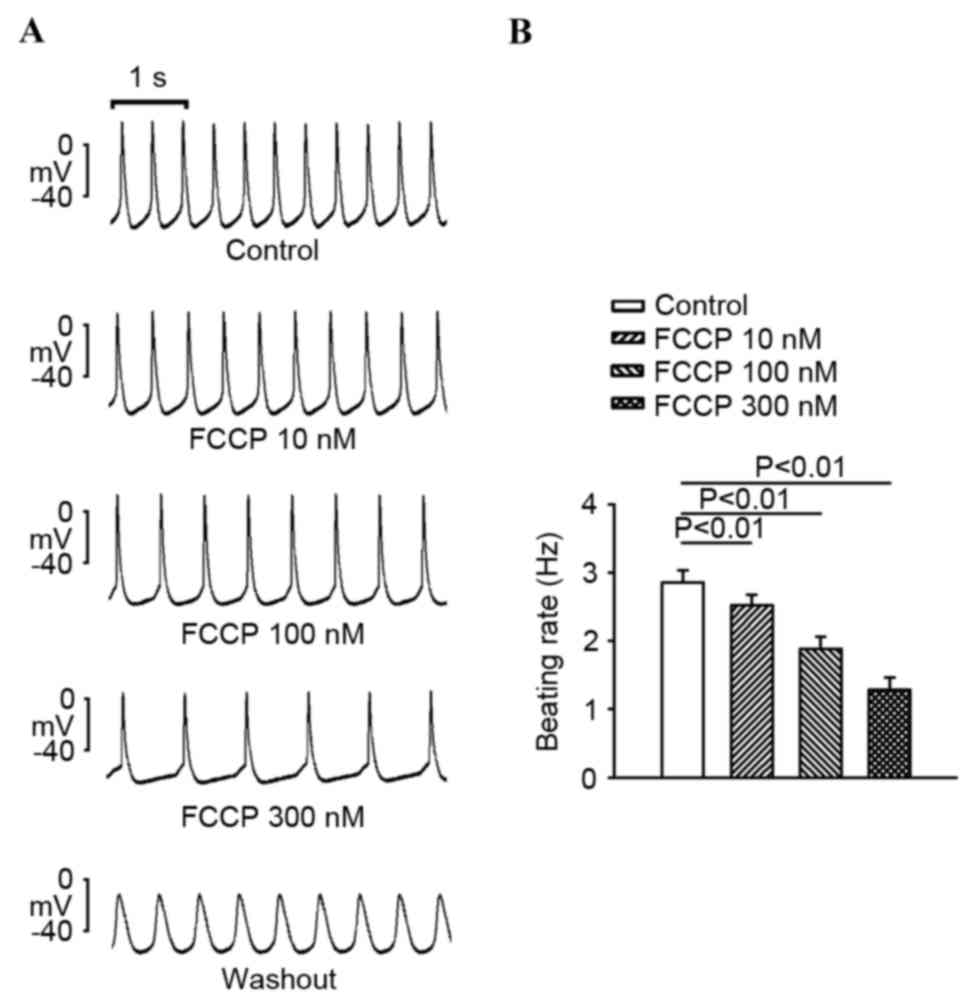
FCCP (10, 100 and 300 nM) significantly decreased
the SAN spontaneous rate in a concentration-dependent manner
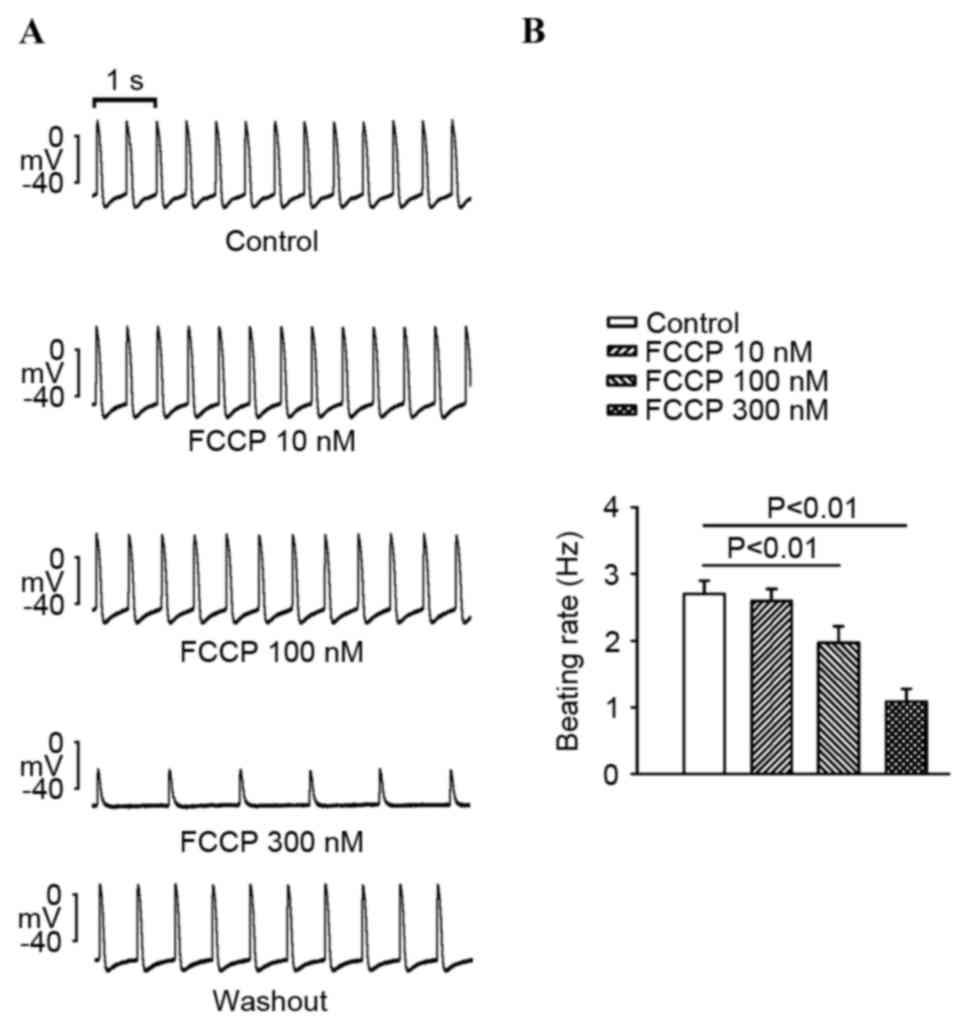
compared with the control (P<0.01; Fig. 1). As exhibited in Fig. 2, FCCP at 100 and 300 nM significantly
decreased PV spontaneous rates compared with the control and FCCP
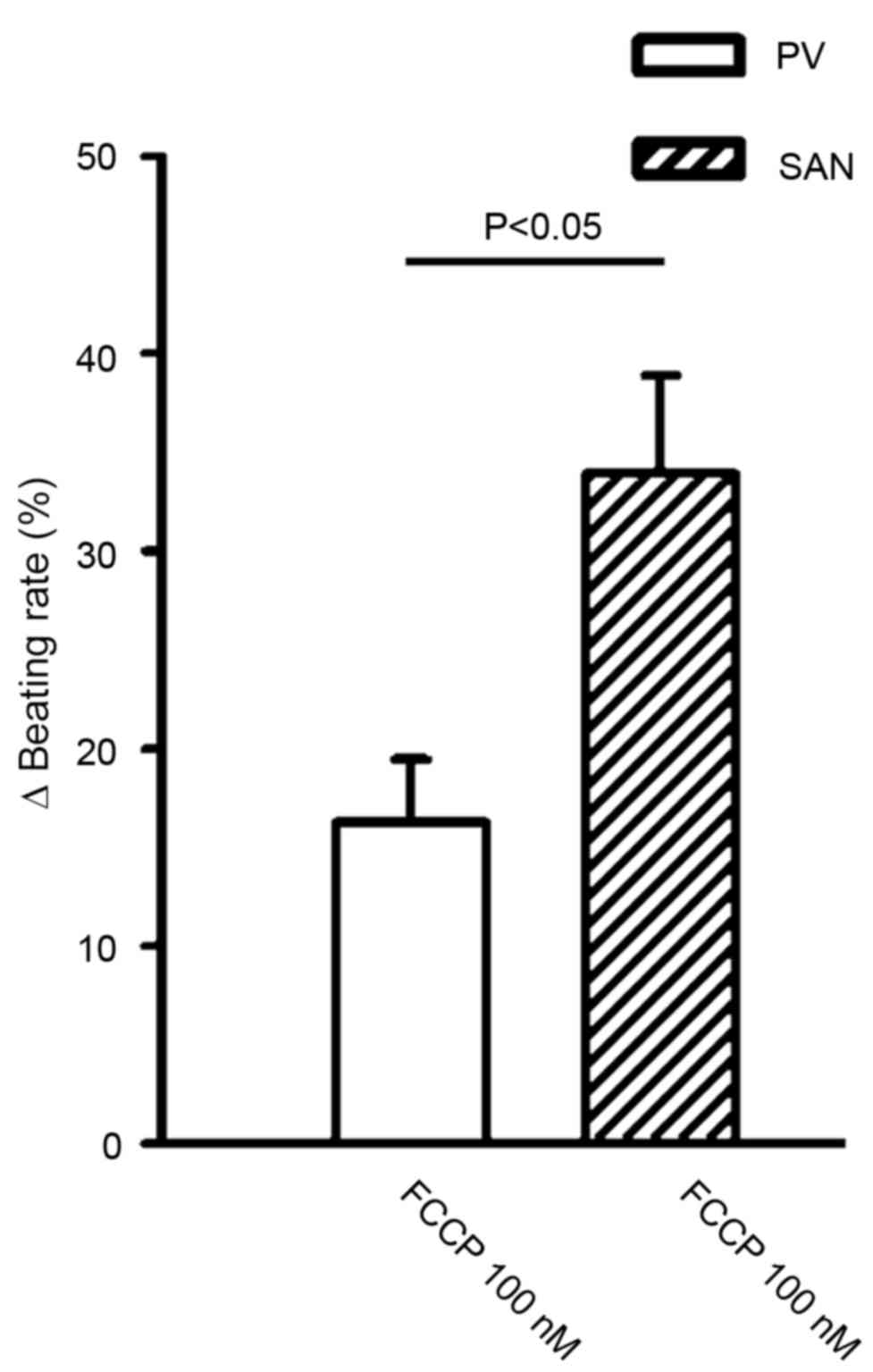
at 10 nM. In addition, FCCP (100 nM) significantly reduced the
beating rate to a greater extent in the SAN than in the PV (34±4.9
vs. 16.3±3.2%; n=6; P<0.05; Fig.
3).
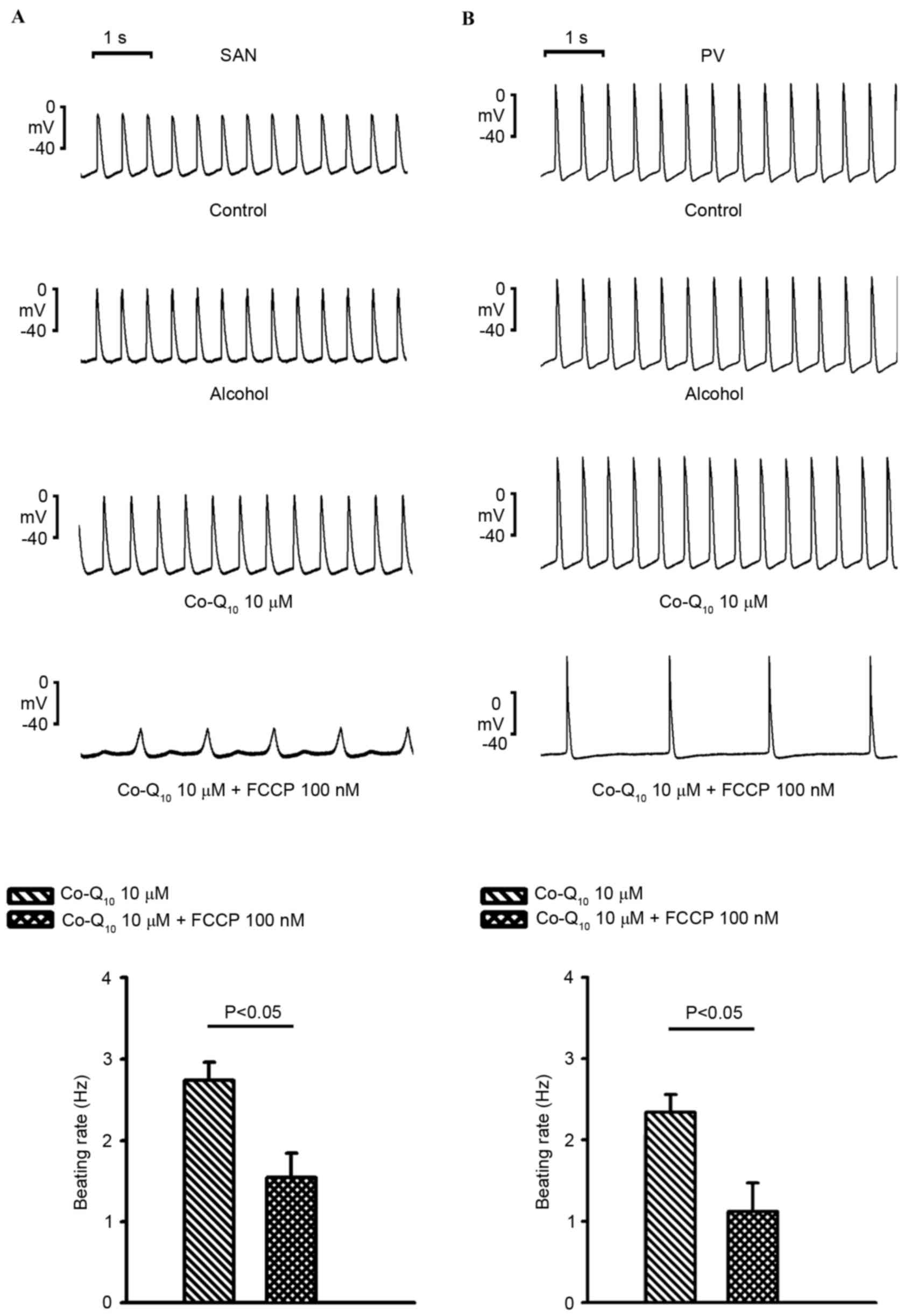
In the presence of Co-Q10 (10 µM), as
exhibited in Fig. 4A and B, FCCP
(100 nM) significantly reduced PV spontaneous beating activity
(2.3±0.2 to 1.1±0.4 Hz; n=5; P<0.05) and SAN spontaneous beating
activity (2.7±0.2 to 1.54±0.3 Hz; n=6; P<0.05) compared with
Co-Q10 alone. In addition, in the presence of
Co-Q10, FCCP (100 nM) reduced the beating rates in the
PV and SVN to a similar extent (51.8±12.7 vs. 41.3±10.5%) compared
with Co-Q10 alone.
Effects of FCCP in the intact PV-SAN
electrical connection
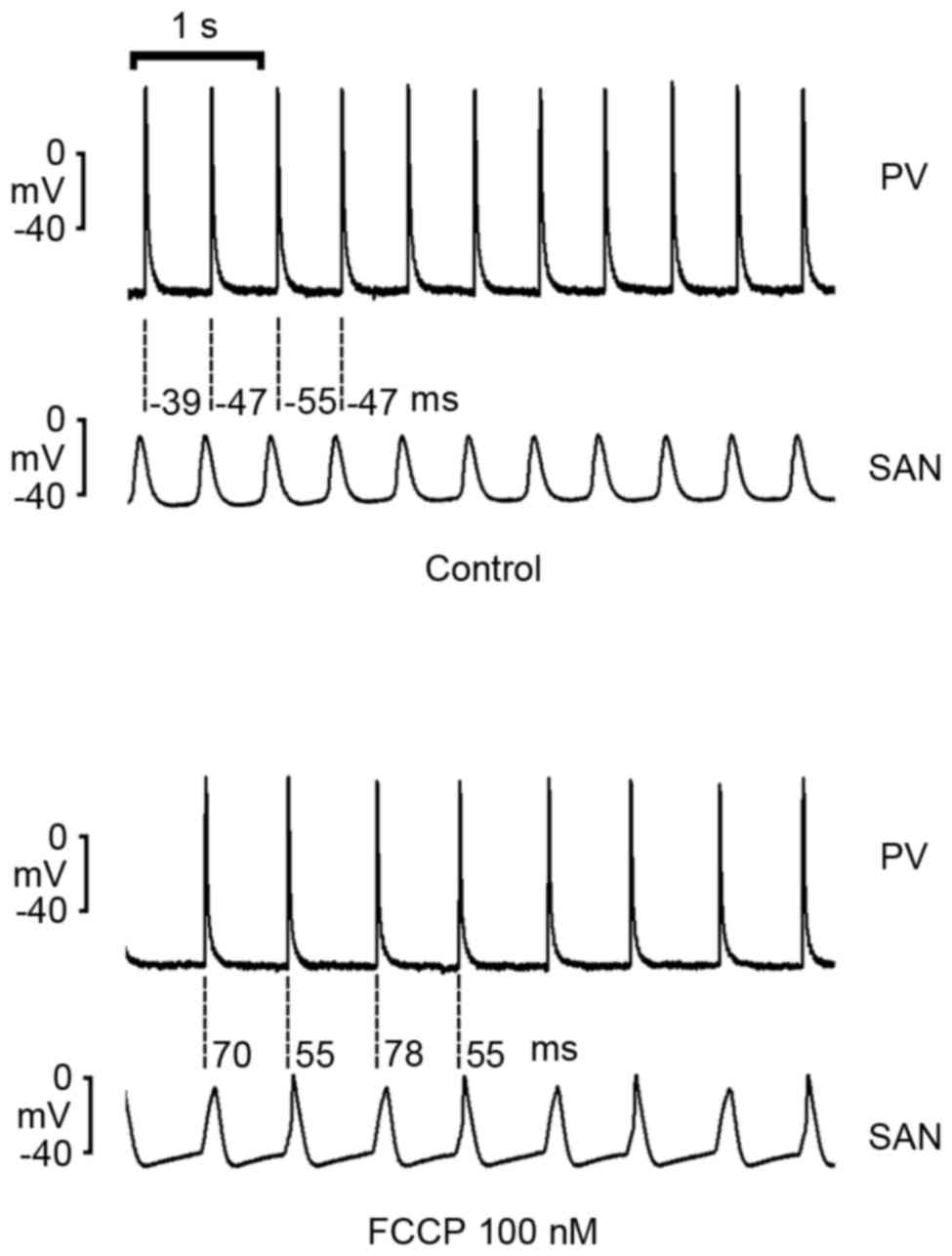
As demonstrated in Fig.
5, FCCP (100 nM) decreased rates in intact SAN-PV preparations;
however, FCCP reversed SAN-to-PV electrical conduction to PV-to-SAN
conduction in 5 of 8 (62.5%) preparations.
Effects of FCCP on the electrical
activities of the RA and LA
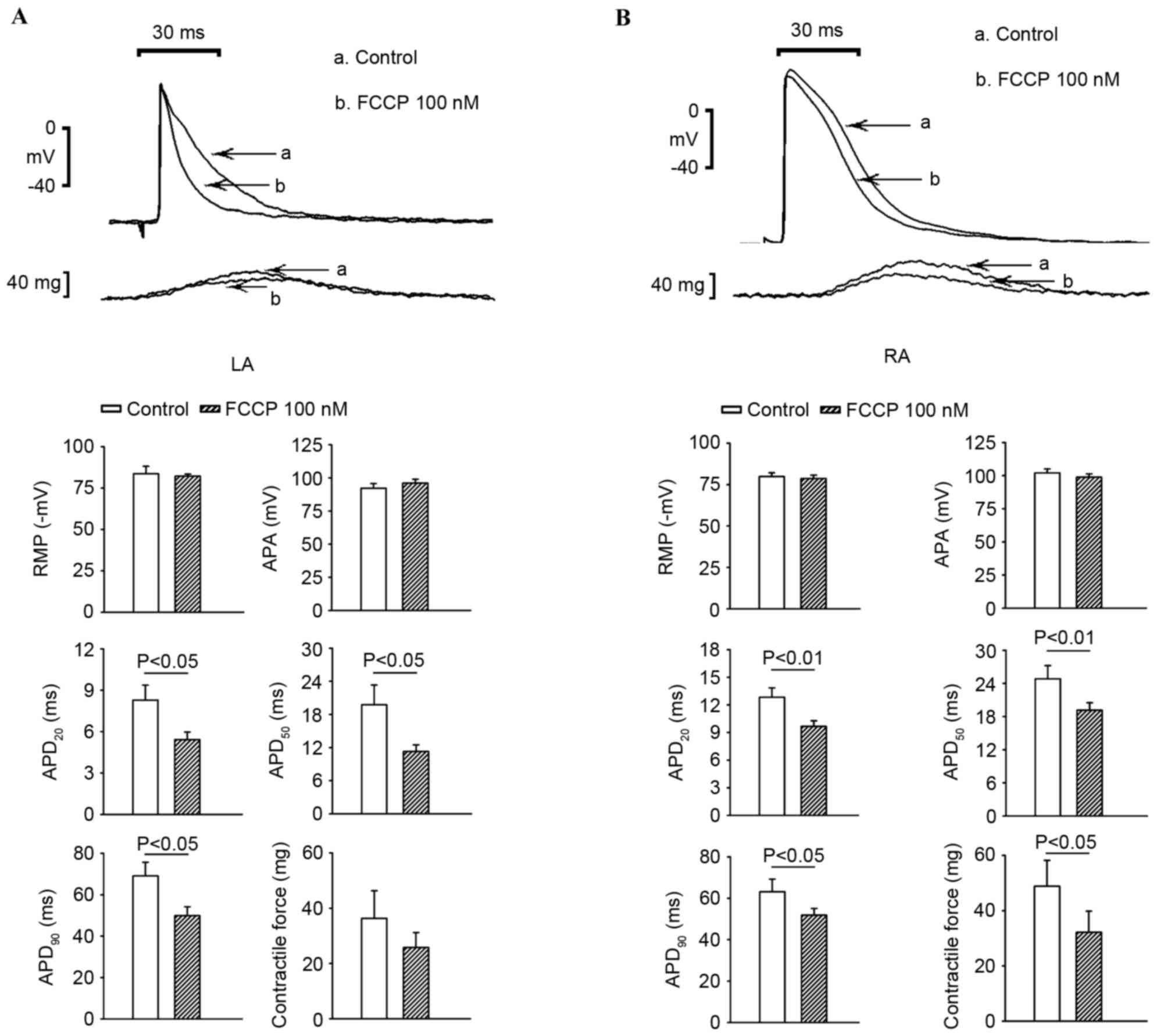
As exhibited in Fig.
6, 100 nM FCCP significantly shortened the APD20,
APD50 and APD90 (P<0.05) and decreased the
contractility in the LA, whereas 100 nM FCCP only shortened the
APD20 to a greater extent in the RA.
Discussion
Cardiac mitochondrial function has important roles
in cardiomyocyte energy metabolism and redox state control, and has
emerged as a target to decrease arrhythmias (6). Hypoxia, which may lead to mitochondrial
dysfunction, has been demonstrated to significantly alter cardiac
electrophysiology (24). In the
present study, it was observed that decreases in PV and SAN
spontaneous activities occurred after FCCP treatment, with a high
probability of reverse overdrive in PV and SAN electrical
interactions. These findings suggest that mitochondrial dysfunction
may modulate PV and SAN electrophysiological properties and enhance
PV arrhythmogenesis through a greater reduction of SAN rates.
Hypoxia is able to decrease the rate of spontaneous
impulse initiation in SAN fibers by decreasing the slope of
diastolic depolarization (25).
Similarly, the present study demonstrated that mitochondrial
dysfunction is able to decrease PV and SAN spontaneous activities.
As mitochondrial dysfunction may lead to an ATP deficiency, the
ATP-sensitive potassium (KATP) channel may subsequently
be influenced and remain open, which may lead to decreasing
pacemaker activity that is noted in hypoxic conditions.
However, in intact PV-SAN preparations, the present
study demonstrated that FCCP (100 nM) altered the electrical
conduction from SAN-to-PV to PV-to-SAN, which may have arisen from
a greater decrease in SAN rates by FCCP with a resulting overdrive
suppression from PVs. This finding suggests an increased
vulnerability of SANs to an ATP deficiency compared with PVs.
Evidence suggests that sinus node dysfunction is able to facilitate
the conditions for AF occurrence by increasing ectopy and
dispersion of refractoriness (23,26,27).
Intact SAN electrical activity is able to suppress arrhythmogenesis
from PVs through a constant overdrive of the PVs. The reverse
overdrive of the PV on the SAN caused by FCCP may facilitate the
occurrence of PV arrhythmogenesis and contribute to mitochondrial
dysfunction-related atrial arrhythmogenesis.
The results of the present study revealed that
Co-Q10 (10 µM) may modulate mitochondrial dysfunction.
The presence of Co-Q10 led to similar FCCP-induced rate
reductions in SANs and PVs, which suggests that the FCCP-induced
PV-overdrive-SAN conduction shift is attenuated by
Co-Q10. A previous study demonstrated that the use of
Co-Q10 as adjuvant treatment in patients with heart
failure may attenuate the incidence of AF (18), which may occur in part through the
protective role of Co-Q10 against mitochondrial
dysfunction-induced PV arrhythmogenesis, as revealed in the present
study. Co-Q10 promoted recovery of ATP following
reoxygenation, which suggests that exogenous Co-Q10 may
facilitate resynthesis of ATP in functionally impaired
mitochondria. Generation of APs in SAN cells is able to be
maintained by a small quantity of ATP (28), which may be produced by exogenous
Co-Q10. A previous study demonstrated that
Co-Q10 did not prevent decreases in ATP in tissues in
the initial period of hypoxia at 30–60 min; however, the ATP
content at 120 min of hypoxia in the presence of Co-Q10
was higher than that of the control (28), which may partially explain the
failure of Co-Q10 to prevent FCCP-induced PV and SAN
rate reductions.
In the present study, FCCP at 100 nM shortened the
APD and decreased contractility slightly in the RA and
significantly in the LA. The influence of the mitochondrial
energetic status on APs is mediated largely by KATP
channels in the membrane. These findings are consistent with
previous studies, whereas hypoxia or ischemia progressively
shortens the APD caused by the opening of KATP channels
(24). Discrepant effects of hypoxia
on AP shortening between the RA and LA were observed in a rabbit
model. Shortening the APD in the RA and LA provides a basis for AF
persistence through facilitating the generation of atrial reentry
circuits. The differential response of the RA and LA to FCCP may
increase dispersions of the APD and may facilitate the maintenance
of AF. Although the mechanisms underlying differences between the
RA and LA are not clear, it is possible that higher expression
levels of heat shock protein 70 in the RA may result in the lower
sensitivity of the RA to FCCP (24).
There were some limitations to the present study.
Firstly, administration of FCCP may produce a non-physiological
condition of mitochondrial dysfunction. Secondly, an acute effect
of mitochondrial dysfunction caused by FCCP application was
observed in the present study, which may differ from the chronic
effect of mitochondrial dysfunction. Finally, the present study
used young, healthy tissue preparations and so results may differ
in pathological settings.
In conclusion, mitochondrial dysfunction regulates
electrophysiological characteristics of the PV, SAN, RA and LA,
which may have a role in the pathophysiology of atrial
arrhythmogenesis.
Acknowledgements
The present study was financially supported by
grants from the Ministry of Science and Technology (grant nos.
MOST103-2314-B-038-041-MY2, MOST103-2314-B-281-005-MY2,
MOST103-2314-B-281-006, MOST103-2314-B-038-055,
NSC102-2314-B-016-029-MY2, NSC102-2325-B-010-005 and
NSC102-2628-B-038-002-MY3), Taipei Medical University (grant no.
TMU101-AE1-B31), Taipei Medical University-Wan Fang Hospital (grant
nos. 101-wf-eva-11, 101-wf-phd-01, 103swf05, 103-wf-eva-02,
104swf02 and 104-wf-eva-01) and Chi-Mei Medical Center (grant nos.
104CM-TMU-07 and CMNDMC10410) of Taiwan.
References
|
1
|
Tsang TS and Gersh BJ: Atrial
fibrillation: An old disease, a new epidemic. Am J Med.
113:432–435. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin YK, Lin FZ, Chen YC, Cheng CC, Lin CI,
Chen YJ and Chen SA: Oxidative stress on pulmonary vein and left
atrium arrhythmogenesis. Circ J. 74:1547–1556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mihm MJ, Yu F, Carnes CA, Reiser PJ,
McCarthy PM, Van Wagoner DR and Bauer JA: Impaired myofibrillar
energetics and oxidative injury during human atrial fibrillation.
Circulation. 104:174–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Takeda S, Mochizuki S, Jindal R
and Dhalla NS: Mechanisms of hydrogen peroxide-induced increase in
intracellular calcium in cardiomyocytes. J Cardiovasc Pharmacol
Ther. 4:41–48. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Wagoner DR: Redox modulation of
cardiac electrical activity. J Cardiovasc Electrophysiol.
12:183–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown DA and O'Rourke B: Cardiac
mitochondria and arrhythmias. Cardiovasc Res. 88:241–249. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeong EM, Liu M, Sturdy M, Gao G, Varghese
ST, Sovari AA and Dudley SC Jr.: Metabolic stress, reactive oxygen
species, and arrhythmia. J Mol Cell Cardiol. 52:454–463. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Montaigne D, Marechal X, Lefebvre P,
Modine T, Fayad G, Dehondt H, Hurt C, Coisne A, Koussa M,
Remy-Jouet I, et al: Mitochondrial dysfunction as an arrhythmogenic
substrate: A translational proof-of-concept study in patients with
metabolic syndrome in whom post-operative atrial fibrillation
develops. J Am Coll Cardiol. 62:1466–1473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ozawa T: Genetic and functional changes in
mitochondria associated with aging. Physiol Rev. 77:425–464.
1997.PubMed/NCBI
|
|
10
|
Wei YH, Lu CY, Lee HC, Pang CY and Ma YS:
Oxidative damage and mutation to mitochondrial DNA and
age-dependent decline of mitochondrial respiratory function. Ann N
Y Acad Sci. 854:155–170. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michikawa Y, Mazzucchelli F, Bresolin N,
Scarlato G and Attardi G: Aging-dependent large accumulation of
point mutations in the human mtDNA control region for replication.
Science. 286:774–779. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang CH, Wu SB, Wu YT and Wei YH:
Oxidative stress response elicited by mitochondrial dysfunction:
Implication in the pathophysiology of aging. Exp Biol Med
(Maywood). 238:450–460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida H, Bao L, Kefaloyianni E, Taskin
E, Okorie U, Hong M, Dhar-Chowdhury P, Kaneko M and Coetzee WA:
AMP-activated protein kinase connects cellular energy metabolism to
KATP channel function. J Mol Cell Cardiol. 52:410–418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sasaki N, Sato T, Marbán E and O'Rourke
B: ATP consumption by uncoupled mitochondria activates sarcolemmal
K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ
Physiol. 280:H1882–H1888. 2001.PubMed/NCBI
|
|
15
|
Liu M, Sanyal S, Gao G, Gurung IS, Zhu X,
Gaconnet G, Kerchner LJ, Shang LL, Huang CL, Grace A, et al:
Cardiac Na+ current regulation by pyridine nucleotides. Circ Res.
105:737–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu M, Liu H and Dudley SC Jr.: Reactive
oxygen species originating from mitochondria regulate the cardiac
sodium channel. Circ Res. 107:967–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matejíková J1, Kucharská J, Pancza D and
Ravingerová T: The effect of antioxidant treatment and NOS
inhibition on the incidence of ischemia-induced arrhythmias in the
diabetic rat heart. Physiol Res. 57 Suppl 2:S55–S60.
2008.PubMed/NCBI
|
|
18
|
Zhao Q, Kebbati AH, Zhang Y, Tang Y,
Okello E and Huang C: Effect of coenzyme Q10 on the incidence of
atrial fibrillation in patients with heart failure. J Investig Med.
63:735–739. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Langsjoen PH and Langsjoen AM: Overview of
the use of CoQ10 in cardiovascular disease. Biofactors. 9:273–284.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Honjo H, Boyett MR, Niwa R, Inada S,
Yamamoto M, Mitsui K, Horiuchi T, Shibata N, Kamiya K and Kodama I:
Pacing-induced spontaneous activity in myocardial sleeves of
pulmonary veins after treatment with ryanodine. Circulation.
107:1937–1943. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen SA, Hsieh MH, Tai CT, Tsai CF,
Prakash VS, Yu WC, Hsu TL, Ding YA and Chang MS: Initiation of
atrial fibrillation by ectopic beats originating from the pulmonary
veins: Electrophysiological characteristics, pharmacological
responses, and effects of radiofrequency ablation. Circulation.
100:1879–1886. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen YJ, Chen SA, Chen YC, Yeh HI, Chan P,
Chang MS and Lin CI: Effects of rapid atrial pacing on the
arrhythmogenic activity of single cardiomyocytes from pulmonary
veins: Implication in initiation of atrial fibrillation.
Circulation. 104:2849–2854. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YC, Lu YY, Cheng CC, Lin YK, Chen SA
and Chen YJ: Sinoatrial node electrical activity modulates
pulmonary vein arrhythmogenesis. Int J Cardiol. 173:447–452. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin YK, Lai MS, Chen YC, Cheng CC, Huang
JH, Chen SA, Chen YJ and Lin CI: Hypoxia and reoxygenation modulate
the arrhythmogenic activity of the pulmonary vein and atrium. Clin
Sci (Lond). 122:121–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Senges J, Mizutani T, Pelzer D, Brachmann
J, Sonnhof U and Kübler W: Effect of hypoxia on the sinoatrial
node, atrium and atrioventricular node in the rabbit heart. Circ
Res. 44:856–863. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luck JC and Engel TR: Dispersion of atrial
refractoriness in patients with sinus node dysfunction.
Circulation. 60:404–412. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loomis TA and Krop S: Auricular
fibrillation induced and maintained in animals by acetylcholine or
vagal stimulation. Circ Res. 3:390–396. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoshikawa Y, Kano T, Higuchi M and Nishi
K: Effects of coenzyme Q10 on recovery of hypoxia-induced changes
in ATP and creatine phosphate contents of sinoatrial nodal cells of
the rabbit's heart after reoxygenation. Arch Int Pharmacodyn Ther.
287:96–108S. 1987.PubMed/NCBI
|