Fn14 was the first widely recognized TWEAK receptor.
Given its one single cysteine-rich domain (CRD), it is generally
considered as the smallest member of the tumor necrosis factor
receptor (TNFR) superfamily (11).
Originally, Fn14 was identified to be induced by fibroblast growth
factor (FGF)1 (22) and subsequent
studies discovered that various other growth factors, including
FGF2, platelet-derived growth factor and epidermal growth factor,
and cytokines such as tumor necrosis factor (TNF)-α, interleukin
(IL)-1β, transforming growth factor-β and interferon-γ, are able to
induce Fn14 expression (22–25).
In contrast to its substantial expression in injured
and morbid tissues, Fn14 is rarely expressed in healthy tissues,
which distinguishes it from TWEAK (26). Studies have indicated that Fn14 is
expressed in the liver following partial hepatectomy and chemical
injury (27,28), in the vasculature following balloon
catheter injury (22), in the kidney
after acute kidney injury and in the heart after myocardial
infarction (29). Regarding the
vascular wall, Fn14 expression is scarce in non-atherosclerotic
arteries whereas TWEAK is typically expressed (15). However, in human atherosclerotic
plaques, both TWEAK and Fn14 are expressed in vascular smooth
muscle cells (VSMCs) and macrophages, suggesting that Fn14 may
interact with TWEAK on damaged vessel walls, which may lead to
adverse outcomes in atherosclerotic lesions (30).
Atherosclerosis is generally considered as a chronic
inflammatory disease with dysfunction of various cells, including
macrophages, smooth muscle cells and endothelial cells (15,31,32). The
interaction between TWEAK and Fn14 may potentially stimulate the
proliferation and migration of cells and induce cell inflammatory
responses, which are critical steps in developing atherosclerotic
plaques (32). The joint function of
TWEAK and Fn14 in disturbing endothelial cells, altering the
phenotypes of smooth muscle cells and triggering the inflammatory
response of monocytes/macrophages, respectively, will be discussed
in subsequent sections.
Normal, healthy endothelium is characterized by its
anticoagulant, antiplatelet and fibrinolytic properties, which are
essential in cardiovascular control (33). However, these functional properties
are easily responsive to stimuli due to the mutable nature of
vascular endothelium. One type of mutation, phenotypic modulation,
may result in dysfunctional state of vascular endothelium, which is
now recognized as the possible initial step in developing
atherosclerosis (33). One important
phenotypic activation biomarker is the expression of adhesion
molecules (15,33). These molecules include selectins,
intercellular adhesion molecules (ICAMs) and vascular adhesion
molecules (VCAMs), which activate and accumulate local monocytes
(15,33). As a result, monocytes are able to
migrate into subendothelial areas and differentiate into
macrophages. These differentiated macrophages are able to uptake
oxidized low density lipoproteins (ox-LDLs).
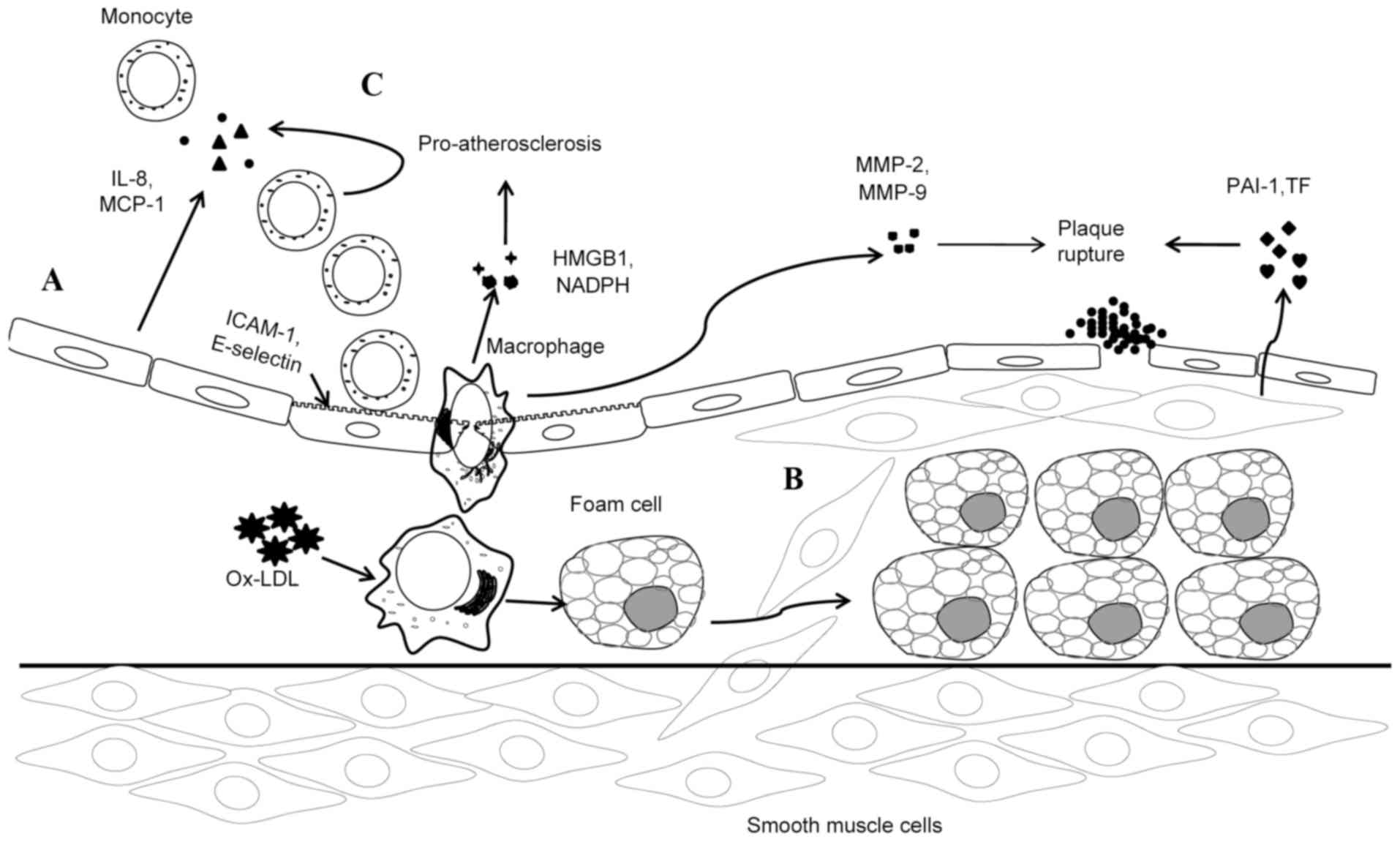
TWEAK induces the expression of adhesion molecules,
such as E-selectin and ICAM-1, on the cell surface of human
umbilical vein endothelial cells (HUVECs), which have been
investigated in vitro (34).
Furthermore, researchers have demonstrated that Fn14 mediates these
processes by blocking monoclonal antibody against human Fn14
(34). TWEAK and Fn14 have been
indicated in HUVECs to jointly induce monocyte chemoattractant
protein-1 (MCP-1) and IL-8 secretion, which predominantly recruits
monocytes and neutrophils (15)
(Fig. 1A).
Plaque rupture or erosion, secondary to complete or
incomplete occlusive thrombosis, reflects the pathophysiological
process of an acute cardiovascular and cerebrovascular event
related to atherosclerosis (14).
Tissue factor (TF), plasminogen activator inhibitor 1 (PAI-1) and
various alternative molecules are responsible for thrombosis, of
which TF is the primary molecule involved in initiating a clotting
cascade (39,40). TFs are identified in cellular pools
in smooth muscle cells (SMCs) and released in microparticles from
SMCs (41). Compared to SMCs in
normal arterial tissue and adjacent media, PAI-1 is increasingly
expressed in SMCs within the fibrous cap (42). This increase in expression is
critical for inhibiting fibrinolysis (43,44). In
human carotid atherosclerotic plaques, Fn14 has been revealed to be
colocalized with both PAI-1 and TF (14). Similarly, in cultured human aortic
SMCs, TWEAK has been observed to induce both TF and PAI-1
expression and activation (14).
Furthermore, with Fn14 small interfering (si)RNA or an anti-Fn14
blocking antibody, the increase in expression and activity of PAI-1
and TF is curbed (Fig. 1B).
Inflammatory response of monocytes/macrophages.
Monocytes/macrophages are involved in atherosclerosis by generating
numerous proinflammatory cytokines (45) and participate in all stages of lesion
development. Assisted by MCP-1 and other chemokines, monocytes are
recruited to endothelium and subsequently, due to high expression
level of endothelial adhesion molecules (such as ICAM-1 and VCAM),
monocytes continue to move into the subendothelial space to
differentiate into macrophages (15,26).
Following this, monocyte-derived macrophages, induced by ox-LDL,
begin to proliferate and extend inflammatory responses (46), thereby increasing the secretion of
proinflammatory cytokines, such as IL-6 and −8, and matrix
metalloproteinases (MMPs), including MMP-9 (31). Production of proinflammatory
cytokines exacerbates the progression of atherosclerosis.
Furthermore, MMPs degrade collagen in the fibrotic cap, resulting
in more-prone-to rupture atherosclerotic plaques (15).
The effects of the TWEAK/Fn14 axis on
monocytes/macrophages have been supported by several studies
(47–49). By inhibiting the biological function
of TWEAK at Fn14 with Fn14-Fc, a study by Schapira et al
(47) demonstrated that TWEAK and
Fn14 interactions may alter macrophage trafficking and increase
lipid uptake of macrophages. Additionally, TWEAK was revealed to
induce various proinflammatory mediators of atherogenesis, such as
IL-6, MCP-1 and IL-8 in activated monocytes (48). There is also evidence that TWEAK is
able to enhance MMP-9 and −2 protease activity in apolipoprotein E
knockout mice (49).
It is well-acknowledged that oxidative stress is
associated with inflammation and the development of atherosclerosis
(50,51). In this scenario, TWEAK assists NADPH
oxidase activation, thereby generating reactive oxygen species
(ROS) in macrophages (12).
Furthermore, NADPH oxidase activity and ROS production are
unsurprisingly compromised by siRNA against Fn14. Therefore,
binding to Fn14 may mediate the role of TWEAK in the context of
oxidative stress (12).
High mobility group box 1 (HMGB1) is a DNA-binding
cytokine released from necrotic cells (52) and activated macrophages (53). HMGB1 stimulates monocytes/macrophages
to express proinflammatory cytokines that mediate the inflammatory
response in atherosclerotic plaque development (54). The TWEAK/Fn14 axis has been
demonstrated to induce monocytes/macrophages to express and secrete
HMGB1 (13). Furthermore, it has
been indicated that both HMGB1 and TWEAK/Fn14 were expressed by
macrophages (55) in human
atherosclerotic plaques (30).
Additionally, a previous study supported this observation in the
shoulder region of human atherosclerotic plaques, where HMGB1 was
colocalized with TWEAK/Fn14 (13).
Therefore, the fact that anti-Fn14 blocking antibody is able to
block the upregulation of HMGB1 by TWEAK in cultured monocytes
supports the notion that Fn14 mediates the functions of TWEAK. In
addition, the same conclusion of HMGB1 expression induced by TWEAK
may be drawn in atherosclerotic plaques of hyperlipidemic
apolipoprotein E-null mice (13)
(Fig. 1C).
CD163 is a 130-kDa member of the scavenger receptors
and is exclusively expressed by monocytes and macrophages (56). CD163 is a marker for
anti-inflammatory activity in macrophages (57) and is potentially a newly identified
receptor for TWEAK, as reported previously (18,21).
Increasing in vivo and in vitro evidence suggests
that the interaction between TWEAK and CD163 may affect the
development of atherosclerosis and related diseases (19,20,57–60).
CD163 is a type I transmembrane protein that belongs
to group B of the scavenger receptor cysteine-rich (SRCR)
superfamily (61). According to the
number of cysteine residues in each SRCR domain, receptors in the
SRCR superfamily are divided into groups A and B (62). CD163 consists of nine consecutive
SRCR class B domains in the extracellular domain and its
transmembrane moiety spans the plasma membrane once (61). In addition, CD163 has a short
intracellular tail that shares consensus sequences for
phosphorylation with protein kinase C and creatine kinase (61). Furthermore, in plasma and body
fluids, such as synovial fluids, CD163 is present in another form,
sCD163, which is a soluble protein (61). Interestingly, sCD163 spans >94% of
the extracellular domain, including all SRCR domains (63).
At present, CD163 is generally considered to be a
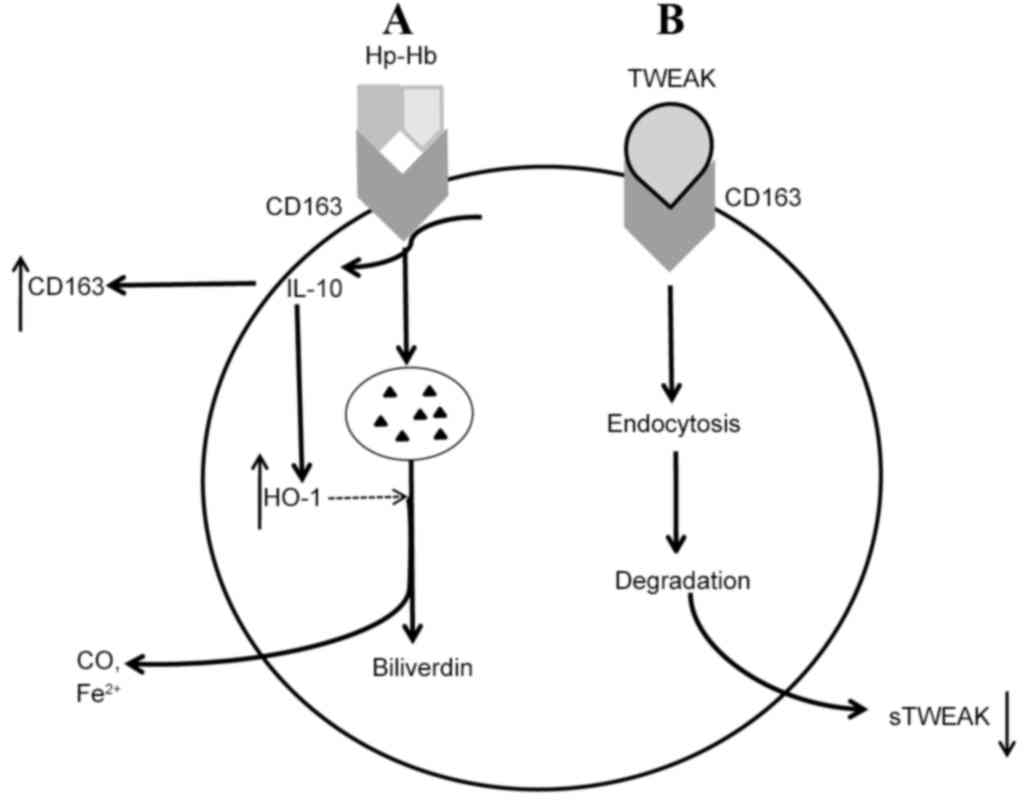
scavenger receptor of Hb-Hp complexes (56), which has been extensively studied.
Intraplaque hemorrhage and subsequent rupture of immature
neointimal vasa vasorum releases cell-free Hb (82,83). Hb
and Hb-derived products are able to enhance oxidative stress and
inflammation, thereby imposing an adverse impact on atherosclerotic
plaque progression (84). However,
Hb may be cleared by CD163. Once released from erythrocytes, during
the process of binding to Hp to form Hb-Hp complexes, Hb exposes a
neo-epitope that is able to combine with CD163 (56,85).
Hb-Hp-CD163 binding accelerates the degradation of the complex
(64). Once the receptor-ligand
complex is delivered to early endosomes through endocytosis, CD163
is dissociated and recycled to cell membrane, whereas Hb-Hp
continues its metabolism within lysosomes (86). Regulated by the rate-limiting enzyme,
heme oxygenase-1 (HO-1), Hb-Hp metabolism produces
anti-inflammatory molecules, including biliverdin, free iron and
carbon monoxide (87,88). Conversely, HO-1 and CD163 production
may be upregulated by IL-10, and production of IL-10 is triggered
by Hb-Hp-CD163 binding (87)
(Fig. 2A).
Besides Hb-Hp complexes, CD163 is also a scavenger
receptor for TWEAK. In 2003, when Fn14 was considered as the
exclusive receptor of TWEAK, a study by Polek et al
(89) indicated that cells lacking
Fn14 were still sensitive to TWEAK, indicating the existence of
another TWEAK receptor. This alternative TWEAK-binding protein was
identified as CD163, via a random combinatorial peptide library
(18). Then, Moreno et al
(21) first reported first reported
the potential role of TWEAK-CD163 binding in atherosclerosis
development. Subsequent research suggested that CD163-expressing
macrophages reduced endogenous TWEAK expression and were able to
bind and degrade TWEAK that was added exogenously from supernatants
in vitro (21). Therefore,
CD163 may inhibit TWEAK, which may further relieve inflammation in
atherosclerosis (90).
The possible contradictory outcomes that TWEAK
exerts when binding to the two different receptors, Fn14 and CD163,
offers a novel therapeutic view in treating atherosclerosis. It is
well-acknowledged that statins are an anti-atherosclerosis
medication that regulate blood lipid metabolism and reduce the
levels of circulating LDL by inhibiting the activity of
hydroxy-methyl-glutaryl Coenzyme A reductase (HMG-CoA reductase), a
key enzyme in the cholesterol biosynthesis pathway (94). Statins, or HMG-CoA reductase
inhibitors, are able to decrease Fn14 expression in human SMCs and
carotid atherosclerotic plaques (30). Furthermore, atorvastatin also reduces
the proinflammatory response induced by TWEAK in human SMCs
(30). Therefore, these findings
indicate that statins may potentially have a novel effect in
atherosclerosis treatment. Additionally, the TWEAK/Fn14 combination
may become a novel target of interference in atherosclerosis
development. As for CD163, in addition to its potential diagnostic
uses in atherosclerosis-related diseases, it is also an interesting
candidate for medical therapy (19,57,58,60).
This is because CD163, as an example of the atheroprotective
intraplaque population, eliminates free-Hb and releases a large
quantity of anti-inflammatory mediators via activating downstream
pathways (62,64). Additionally, CD163 has the potential
to diminish inflammation in atherosclerosis by recognizing and
internalizing TWEAK.
In conclusion, TWEAK functions divergently when
binding to the two receptors, Fn14 and CD163. Regarding Fn14, TWEAK
imposes adverse effects by encouraging atherosclerosis development,
including disturbing endothelial cells, changing phenotypes of
smooth muscle cells and triggering inflammatory response of
monocytes/macrophages. However, when interacting with CD163, TWEAK
may elicit potential protective effects. Currently, the mechanism
by which the downstream pathway is activated following the
recognition and internalization of TWEAK by CD163 and whether this
activated pathway is similar to the result of CD163 binding to the
Hp-Hb complex is unclear. Further study in vivo is required
to elucidate the effect of the TWEAK/CD163 axis.
The present study was supported by funding from the
National Basic Research Program of China (grant no. 2014CB542400),
the National Natural Science Foundation of China (grant nos.
81470593 and 81300053) and the Research Innovation Program for
Graduate Students of Central South University (grant no.
2016zzts152).
|
1
|
Linden F, Domschke G, Erbel C, Akhavanpoor
M, Katus HA and Gleissner CA: Inflammatory therapeutic targets in
coronary atherosclerosis-from molecular biology to clinical
application. Front Physiol. 5:4552014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Scott J: The pathogenesis of
atherosclerosis and new opportunities for treatment and prevention.
J Neural Transm Suppl. 1–17. 2002.PubMed/NCBI
|
|
3
|
Tedgui A and Mallat Z: Cytokines in
atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev.
86:515–581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blanco-Colio LM, Martin-Ventura JL,
Carrero JJ, Yilmaz MI, Moreno JA, Gómez-Guerrero C, Ortiz A and
Egido J: Vascular proteomics and the discovery process of clinical
biomarkers: The case of TWEAK. Proteomics Clin Appl. 5:281–288.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jelic-Ivanović Z, Bujisić N, Spasić S,
Bogavac-Stanojević N, Spasojević-Kalimanovska V and
Kotur-Stevuljević J: Circulating sTWEAK improves the prediction of
coronary artery disease. Clin Biochem. 42:1381–1386. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kralisch S, Ziegelmeier M, Bachmann A,
Seeger J, Lössner U, Blüher M, Stumvoll M and Fasshauer M: Serum
levels of the atherosclerosis biomarker sTWEAK are decreased in
type 2 diabetes and end-stage renal disease. Atherosclerosis.
199:440–444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Blanco-Colio LM, Martín-Ventura JL,
Muñóz-García B, Orbe J, Páramo JA, Michel JB, Ortiz A, Meilhac O
and Egido J: Identification of soluble tumor necrosis factor-like
weak inducer of apoptosis (sTWEAK) as a possible biomarker of
subclinical atherosclerosis. Arterioscler Thromb Vasc Biol.
27:916–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Munoz-García B, Moreno JA, López-Franco O,
Sanz AB, Martín-Ventura JL, Blanco J, Jakubowski A, Burkly LC,
Ortiz A, Egido J and Blanco-Colio LM: Tumor necrosis factor-like
weak inducer of apoptosis (TWEAK) enhances vascular and renal
damage induced by hyperlipidemic diet in ApoE-knockout mice.
Arterioscler Thromb Vasc Biol. 29:2061–2068. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chicheportiche Y, Bourdon PR, Xu H, Hsu
YM, Scott H, Hession C, Garcia I and Browning JL: TWEAK, a new
secreted ligand in the tumor necrosis factor family that weakly
induces apoptosis. J Biol Chem. 272:32401–32410. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bodmer JL, Schneider P and Tschopp J: The
molecular architecture of the TNF superfamily. Trends Biochem Sci.
27:19–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiley SR, Cassiano L, Lofton T,
Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA
and Fanslow WC: A novel TNF receptor family member binds TWEAK and
is implicated in angiogenesis. Immunity. 15:837–846. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Madrigal-Matute J, Fernandez-Laso V,
Sastre C, Llamas-Granda P, Egido J, Martin-Ventura JL, Zalba G and
Blanco-Colio LM: TWEAK/Fn14 interaction promotes oxidative stress
through NADPH oxidase activation in macrophages. Cardiovasc Res.
108:139–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moreno JA, Sastre C, Madrigal-Matute J,
Muñoz-García B, Ortega L, Burkly LC, Egido J, Martín-Ventura JL and
Blanco-Colio LM: HMGB1 expression and secretion are increased via
TWEAK-Fn14 interaction in atherosclerotic plaques and cultured
monocytes. Arterioscler Thromb Vasc Biol. 33:612–620. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Muñoz-Garcia B, Madrigal-Matute J, Moreno
JA, Martin-Ventura JL, López-Franco O, Sastre C, Ortega L, Burkly
LC, Egido J and Blanco-Colio LM: TWEAK-Fn14 interaction enhances
plasminogen activator inhibitor 1 and tissue factor expression in
atherosclerotic plaques and in cultured vascular smooth muscle
cells. Cardiovasc Res. 89:225–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Blanco-Colio LM, Martín-Ventura JL,
Munoz-Garcia B, Moreno JA, Meilhac O, Ortiz A and Egido J: TWEAK
and Fn14. New players in the pathogenesis of atherosclerosis. Front
Biosci. 12:3648–3655. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakayama M, Harada N, Okumura K and Yagita
H: Characterization of murine TWEAK and its receptor (Fn14) by
monoclonal antibodies. Biochem Biophys Res Commun. 306:819–825.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wiley SR and Winkles JA: TWEAK, a member
of the TNF superfamily, is a multifunctional cytokine that binds
the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 14:241–249.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bover LC, Cardó-Vila M, Kuniyasu A, Sun J,
Rangel R, Takeya M, Aggarwal BB, Arap W and Pasqualini R: A
previously unrecognized protein-protein interaction between TWEAK
and CD163: Potential biological implications. J Immunol.
178:8183–8194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ilter A, Orem C, Yucesan F Balaban, Sahin
M, Hosoglu Y, Kurumahmutoglu E, Yaman S Ozer and Orem A: Evaluation
of serum sTWEAK and sCD163 levels in patients with acute and
chronic coronary artery disease. Int J Clin Exp Med. 8:9394–9402.
2015.PubMed/NCBI
|
|
20
|
Valdivielso JM, Coll B, Martin-Ventura JL,
Moreno JA, Egido J, Fernández E and Blanco-Colio LM: Soluble TWEAK
is associated with atherosclerotic burden in patients with chronic
kidney disease. J Nephrol Nephrol Nephrol. 26:1105–1113. 2013.
|
|
21
|
Moreno JA, Muñoz-García B, Martín-Ventura
JL, Madrigal-Matute J, Orbe J, Páramo JA, Ortega L, Egido J and
Blanco-Colio LM: The CD163-expressing macrophages recognize and
internalize TWEAK Potential consequences in atherosclerosis.
Atherosclerosis. 207:103–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng SL, Guo Y, Factor VM, Thorgeirsson
SS, Bell DW, Testa JR, Peifley KA and Winkles JA: The Fn14
immediate-early response gene is induced during liver regeneration
and highly expressed in both human and murine hepatocellular
carcinomas. Am J Pathol. 156:1253–1261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meighan-Mantha RL, Hsu DK, Guo Y, Brown
SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ,
Jenkins NA, et al: The mitogen-inducible Fn14 gene encodes a type I
transmembrane protein that modulates fibroblast adhesion and
migration. J Biol Chem. 274:33166–33176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brown SA, Richards CM, Hanscom HN, Feng SL
and Winkles JA: The Fn14 cytoplasmic tail binds
tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5
and mediates nuclear factor-kappaB activation. Biochem J.
371:395–403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saitoh T, Nakayama M, Nakano H, Yagita H,
Yamamoto N and Yamaoka S: TWEAK induces NF-kappaB2 p100 processing
and long lasting NF-kappaB activation. J Biol Chem.
278:36005–36012. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blanco-Colio LM: TWEAK/Fn14 Axis: A
promising target for the treatment of cardiovascular diseases.
Front Immunol. 5:32014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Felli N, Pedini F, Zeuner A, Petrucci E,
Testa U, Conticello C, Biffoni M, Di Cataldo A, Winkles JA, Peschle
C and De Maria R: Multiple members of the TNF superfamily
contribute to IFN-gamma-mediated inhibition of erythropoiesis. J
Immunol. 175:1464–1472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jakubowski A, Browning B, Lukashev M,
Sizing I, Thompson JS, Benjamin CD, Hsu YM, Ambrose C, Zheng TS and
Burkly LC: Dual role for TWEAK in angiogenic regulation. J Cell
Sci. 115:267–274. 2002.PubMed/NCBI
|
|
29
|
Justo P, Sanz AB, Sanchez-Niño MD, Winkles
JA, Lorz C, Egido J and Ortiz A: Cytokine cooperation in renal
tubular cell injury: The role of TWEAK. Kidney Int. 70:1750–1758.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Munoz-García B, Martin-Ventura JL,
Martínez E, Sánchez S, Hernández G, Ortega L, Ortiz A, Egido J and
Blanco-Colio LM: Fn14 is upregulated in cytokine-stimulated
vascular smooth muscle cells and is expressed in human carotid
atherosclerotic plaques: Modulation by atorvastatin. Stroke.
37:2044–2053. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tuttolomondo A, Di Raimondo D, Pecoraro R,
Arnao V, Pinto A and Licata G: Atherosclerosis as an inflammatory
disease. Curr Pharm Des. 18:4266–4288. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weissberg PL and Bennett MR:
Atherosclerosis-an inflammatory disease. N Engl J Med.
340:1928–1929. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gimbrone MA Jr and Garcia-Cardeña G:
Vascular endothelium, hemodynamics, and the pathobiology of
atherosclerosis. Cardiovasc Pathol. 22:9–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harada N, Nakayama M, Nakano H, Fukuchi Y,
Yagita H and Okumura K: Pro-inflammatory effect of TWEAK/Fn14
interaction on human umbilical vein endothelial cells. Biochem
Biophys Res Commun. 299:488–493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi N and Chen SY: Mechanisms
simultaneously regulate smooth muscle proliferation and
differentiation. J Biomed Res. 28:40–46. 2014.PubMed/NCBI
|
|
36
|
Chistiakov DA, Orekhov AN and Bobryshev
YV: Vascular smooth muscle cell in atherosclerosis. Acta Physiol
(Oxf). 214:33–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lynch CN, Wang YC, Lund JK, Chen YW, Leal
JA and Wiley SR: TWEAK induces angiogenesis and proliferation of
endothelial cells. J Biol Chem. 274:8455–8459. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han S, Yoon K, Lee K, Kim K, Jang H, Lee
NK, Hwang K and Lee S Young: TNF-related weak inducer of apoptosis
receptor, a TNF receptor superfamily member, activates NF-kappa B
through TNF receptor-associated factors. Biochem Biophys Res
Commun. 305:789–796. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mackman N: Role of tissue factor in
hemostasis and thrombosis. Blood Cells Mol Dis. 36:104–107. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Potempa J, Korzus E and Travis J: The
serpin superfamily of proteinase inhibitors: Structure, function,
and regulation. J Biol Chem. 269:15957–15960. 1994.PubMed/NCBI
|
|
41
|
Schecter AD, Spirn B, Rossikhina M, Giesen
PL, Bogdanov V, Fallon JT, Fisher EA, Schnapp LM, Nemerson Y and
Taubman MB: Release of active tissue factor by human arterial
smooth muscle cells. Circ Res. 87:126–132. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lupu F, Bergonzelli GE, Heim DA, Cousin E,
Genton CY, Bachmann F and Kruithof EK: Localization and production
of plasminogen activator inhibitor-1 in human healthy and
atherosclerotic arteries. Arterioscler Thromb. 13:1090–1100. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Taubman MB, Fallon JT, Schecter AD, Giesen
P, Mendlowitz M, Fyfe BS, Marmur JD and Nemerson Y: Tissue factor
in the pathogenesis of atherosclerosis. Thromb Haemost. 78:200–204.
1997.PubMed/NCBI
|
|
44
|
Agirbasli M: Pivotal role of
plasminogen-activator inhibitor 1 in vascular disease. Int J Clin
Pract. 59:102–106. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Spinas E, Kritas SK, Saggini A, Mobili A,
Caraffa A, Antinolfi P, Pantalone A, Tei M, Speziali A, Saggini R
and Conti P: Role of mast cells in atherosclerosis: A classical
inflammatory disease. Int J Immunopathol Pharmacol. 27:517–521.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hansson GK: Immune mechanisms in
atherosclerosis. Arterioscler Thromb Vasc Biol. 21:1876–1890. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schapira K, Burkly LC, Zheng TS, Wu P,
Groeneweg M, Rousch M, Kockx MM, Daemen MJ and Heeneman S: Fn14-Fc
fusion protein regulates atherosclerosis in ApoE-/-mice and
inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc
Biol. 29:2021–2027. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim SH, Kang YJ, Kim WJ, Woo DK, Lee Y,
Kim DI, Park YB, Kwon BS, Park JE and Lee WH: TWEAK can induce
pro-inflammatory cytokines and matrix metalloproteinase-9 in
macrophages. Circ J. 68:396–399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sastre C, Fernández-Laso V,
Madrigal-Matute J, Muñoz-García B, Moreno JA, Pastor-Vargas C,
Llamas-Granda P, Burkly LC, Egido J, Martín-Ventura JL and
Blanco-Colio LM: Genetic deletion or TWEAK blocking antibody
administration reduce atherosclerosis and enhance plaque stability
in mice. J Cell Mol Med Med Cell Mol Med. 18:721–734. 2014.
View Article : Google Scholar
|
|
50
|
Madamanchi NR and Runge MS: Mitochondrial
dysfunction in atherosclerosis. Circ Res. 100:460–473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Munzel T, Gori T, Bruno RM and Taddei S:
Is oxidative stress a therapeutic target in cardiovascular disease?
Eur Heart J. 31:2741–2748. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kalinina N, Agrotis A, Antropova Y,
DiVitto G, Kanellakis P, Kostolias G, Ilyinskaya O, Tararak E and
Bobik A: Increased expression of the DNA-binding cytokine HMGB1 in
human atherosclerotic lesions: Role of activated macrophages and
cytokines. Arterioscler Thromb Vasc Biol. 24:2320–2325. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Andersson U, Erlandsson-Harris H, Yang H
and Tracey KJ: HMGB1 as a DNA-binding cytokine. J Leukoc Biol.
72:1084–1091. 2002.PubMed/NCBI
|
|
55
|
Inoue K, Kawahara K, Biswas KK, Ando K,
Mitsudo K, Nobuyoshi M and Maruyama I: HMGB1 expression by
activated vascular smooth muscle cells in advanced human
atherosclerosis plaques. Cardiovasc Patho. 16:136–143. 2007.
View Article : Google Scholar
|
|
56
|
Kristiansen M, Graversen JH, Jacobsen C,
Sonne O, Hoffman HJ, Law SK and Moestrup SK: Identification of the
haemoglobin scavenger receptor. Nature. 409:198–201. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jasiewicz M, Kowal K, Kowal-Bielecka O,
Knapp M, Skiepko R, Bodzenta-Lukaszyk A, Sobkowicz B, Musial WJ and
Kaminski KA: Serum levels of CD163 and TWEAK in patients with
pulmonary arterial hypertension. Cytokine. 66:40–45. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Urbonaviciene G, Martin-Ventura JL,
Lindholt JS, Urbonavicius S, Moreno JA, Egido J and Blanco-Colio
LM: Impact of soluble TWEAK and CD163/TWEAK ratio on long-term
cardiovascular mortality in patients with peripheral arterial
disease. Atherosclerosis. 219:892–899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Beltran LM, Muñoz Hernández R, de Pablo
Bernal RS, García Morillo JS, Egido J, Noval ML, Ferrando-Martinez
S, Blanco-Colio LM, Genebat M, Villar JR, et al: Reduced sTWEAK and
increased sCD163 levels in HIV-infected patients: Modulation by
antiretroviral treatment, HIV replication and HCV co-infection.
PLoS One. 9:e905412014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Llauradó G, González-Clemente JM,
Maymó-Masip E, Subías D, Vendrell J and Chacón MR: Serum levels of
TWEAK and scavenger receptor CD163 in type 1 diabetes mellitus:
Relationship with cardiovascular risk factors. A case-control
study. PLoS One. 7:e439192012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Van Gorp H, Delputte PL and Nauwynck HJ:
Scavenger receptor CD163, a Jack-of-all-trades and potential target
for cell-directed therapy. Mol Immunol. 47:1650–1660. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kowal K, Silver R, Sławińska E, Bielecki
M, Chyczewski L and Kowal-Bielecka O: CD163 and its role in
inflammation. Folia Histochem Cytobiol. 49:365–374. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Moller HJ, Nielsen MJ, Maniecki MB, Madsen
M and Moestrup SK: Soluble macrophage-derived CD163: A homogenous
ectodomain protein with a dissociable haptoglobin-hemoglobin
binding. Immunobiology. 215:406–412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Etzerodt A and Moestrup SK: CD163 and
inflammation: Biological, diagnostic, and therapeutic aspects.
Antioxid Redox Signal. 18:2352–2363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Backe E, Schwarting R, Gerdes J, Ernst M
and Stein H: Ber-MAC3: New monoclonal antibody that defines human
monocyte/macrophage differentiation antigen. J Clin Pathol.
44:936–945. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chamorro S, Revilla C, Alvarez B, Alonso
F, Ezquerra A and Domínguez J: Phenotypic and functional
heterogeneity of porcine blood monocytes and its relation with
maturation. Immunology. 114:63–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Baeten D, Møller HJ, Delanghe J, Veys EM,
Moestrup SK and De Keyser F: Association of CD163+ macrophages and
local production of soluble CD163 with decreased lymphocyte
activation in spondylarthropathy synovitis. Arthritis Rheum.
50:1611–1623. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bronkhorst IH, Ly LV, Jordanova ES,
Vrolijk J, Versluis M, Luyten GP and Jager MJ: Detection of
M2-macrophages in uveal melanoma and relation with survival. Invest
Ophthalmol Vis Sci. 52:643–650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ratcliffe NR, Kennedy SM and Morganelli
PM: Immunocytochemical detection of Fcgamma receptors in human
atherosclerotic lesions. Immunol Lett. 77:169–174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Högger P, Dreier J, Droste A, Buck F and
Sorg C: Identification of the integral membrane protein RM3/1 on
human monocytes as a glucocorticoid-inducible member of the
scavenger receptor cysteine-rich family (CD163). J Immunol.
161:1883–1890. 1998.PubMed/NCBI
|
|
71
|
Sulahian TH, Högger P, Wahner AE, Wardwell
K, Goulding NJ, Sorg C, Droste A, Stehling M, Wallace PK,
Morganelli PM and Guyre PM: Human monocytes express CD163, which is
upregulated by IL-10 and identical to p155. Cytokine. 12:1312–1321.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Timmermann M and Högger P: Oxidative
stress and 8-iso-prostaglandin F(2alpha) induce ectodomain shedding
of CD163 and release of tumor necrosis factor-alpha from human
monocytes. Free Radic Biol Med. 39:98–107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zwadlo G, Voegeli R, Osthoff K Schulze and
Sorg C: A monoclonal antibody to a novel differentiation antigen on
human macrophages associated with the down-regulatory phase of the
inflammatory process. Exp Cell Biol. 55:295–304. 1987.PubMed/NCBI
|
|
74
|
Buechler C, Ritter M, Orsó E, Langmann T,
Klucken J and Schmitz G: Regulation of scavenger receptor CD163
expression in human monocytes and macrophages by pro- and
antiinflammatory stimuli. J Leukoc Biol. 67:97–103. 2000.PubMed/NCBI
|
|
75
|
Van den Heuvel MM, Tensen CP, van As JH,
Van den Berg TK, Fluitsma DM, Dijkstra CD, Döpp EA, Droste A, Van
Gaalen FA, Sorg C, et al: Regulation of CD 163 on human
macrophages: Cross-linking of CD163 induces signaling and
activation. J Leukoc Biol. 66:858–866. 1999.PubMed/NCBI
|
|
76
|
Schaer DJ, Boretti FS, Schoedon G and
Schaffner A: Induction of the CD163-dependent haemoglobin uptake by
macrophages as a novel anti-inflammatory action of glucocorticoids.
Br J Haematol. 119:239–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Varga G, Ehrchen J, Tsianakas A, Tenbrock
K, Rattenholl A, Seeliger S, Mack M, Roth J and Sunderkoetter C:
Glucocorticoids induce an activated, anti-inflammatory monocyte
subset in mice that resembles myeloid-derived suppressor cells. J
Leukoc Biol. 84:644–650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hogger P, Erpenstein U, Rohdewald P and
Sorg C: Biochemical characterization of a glucocorticoid-induced
membrane protein (RM3/1) in human monocytes and its application as
model system for ranking glucocorticoid potency. Pharm Res.
15:296–302. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Weaver LK, Pioli PA, Wardwell K, Vogel SN
and Guyre PM: Up-regulation of human monocyte CD163 upon activation
of cell-surface Toll-like receptors. J Leukoc Biol. 81:663–671.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hintz KA, Rassias AJ, Wardwell K, Moss ML,
Morganelli PM, Pioli PA, Givan AL, Wallace PK, Yeager MP and Guyre
PM: Endotoxin induces rapid metalloproteinase-mediated shedding
followed by up-regulation of the monocyte hemoglobin scavenger
receptor CD163. J Leukoc Biol. 72:711–717. 2002.PubMed/NCBI
|
|
81
|
Gleissner CA, Shaked I, Erbel C, Böckler
D, Katus HA and Ley K: CXCL4 downregulates the atheroprotective
hemoglobin receptor CD163 in human macrophages. Circ Res.
106:203–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Levy AP and Moreno PR: Intraplaque
hemorrhage. Curr Mol Med. 6:479–488. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Virmani R, Kolodgie FD, Burke AP, Finn AV,
Gold HK, Tulenko TN, Wrenn SP and Narula J: Atherosclerotic plaque
progression and vulnerability to rupture: Angiogenesis as a source
of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol.
25:2054–2061. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Schaer DJ and Buehler PW: Cell-free
hemoglobin and its scavenger proteins: New disease models leading
the way to targeted therapies. Cold Spring Harb Perspect Med.
3(pii): a0134332013.PubMed/NCBI
|
|
85
|
Madsen M, Møller HJ, Nielsen MJ, Jacobsen
C, Graversen JH, van den Berg T and Moestrup SK: Molecular
characterization of the haptoglobin.hemoglobin receptor CD163.
Ligand binding properties of the scavenger receptor cysteine-rich
domain region. J Biol Chem. 279:51561–51567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Schaer CA, Schoedon G, Imhof A, Kurrer MO
and Schaer DJ: Constitutive endocytosis of CD163 mediates
hemoglobin-heme uptake and determines the noninflammatory and
protective transcriptional response of macrophages to hemoglobin.
Circ Res. 99:943–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Philippidis P, Mason JC, Evans BJ, Nadra
I, Taylor KM, Haskard DO and Landis RC: Hemoglobin scavenger
receptor CD163 mediates interleukin-10 release and heme oxygenase-1
synthesis: Antiinflammatory monocyte-macrophage responses in vitro,
in resolving skin blisters in vivo, and after cardiopulmonary
bypass surgery. Circ Res. 94:119–126. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Nielsen MJ, Møller HJ and Moestrup SK:
Hemoglobin and heme scavenger receptors. Antioxid Redox Signal.
12:261–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Polek TC, Talpaz M, Darnay BG and
Spivak-Kroizman T: TWEAK mediates signal transduction and
differentiation of RAW264.7 cells in the absence of Fn14/TweakR.
Evidence for a second TWEAK receptor. J Biol Chem. 278:32317–32323.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Burkly LC, Michaelson JS, Hahm K,
Jakubowski A and Zheng TS: TWEAKing tissue remodeling by a
multifunctional cytokine: Role of TWEAK/Fn14 pathway in health and
disease. Cytokine. 40:1–16. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Moreno JA, Dejouvencel T, Labreuche J,
Smadja DM, Dussiot M, Martin-Ventura JL, Egido J, Gaussem P,
Emmerich J, Michel JB, et al: Peripheral artery disease is
associated with a high CD163/TWEAK plasma ratio. Arterioscler
Thromb Vasc Biol. 30:1253–1262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kowal-Bielecka O, Bielecki M, Guiducci S,
Trzcinska-Butkiewicz B, Michalska-Jakubus M, Matucci-Cerinic M,
Brzosko M, Krasowska D, Chyczewski L and Kowal K: High serum
sCD163/sTWEAK ratio is associated with lower risk of digital ulcers
but more severe skin disease in patients with systemic sclerosis.
Arthritis Res Ther. 15:R692013. View
Article : Google Scholar : PubMed/NCBI
|
|
93
|
Møller HJ: Soluble CD163. Scand J Clin Lab
Invest. 72:1–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|