Introduction
Head and neck cancer is the sixth most prevalent
malignancy and is increasing in frequency worldwide (1). The most common site for head and neck
cancer is the oral cavity, with 40% of cases occurring in this
region (2). Despite improvements in
surgery, radiation and chemotherapy, treatment of oral squamous
cell carcinoma (OSCC) remains a challenge. Although OSCC has been
extensively studied (3), the
molecular characteristics of this malignancy remain unknown.
A decrease in epithelial cell adhesion is a key step
in the progression and metastasis of tumors (4). The loss of epithelial differentiation
and gain of a mesenchymal phenotype, known as
epithelial-to-mesenchymal transition, is associated with malignant
transformation in numerous carcinomas (5), and may be a predictor of OSCC
progression and prognosis (6,7).
The epithelial junctional complex is composed of
tight junctions, adherens junctions and desmosomes. Cadherins are
major components of adherens junctions and serve a key role in the
maintenance of epithelial tissue integrity (8). The transmembrane protein epithelial
(E)-cadherin is a widely distributed, intercelluar adhesion
molecule (9) that, through its
cytoplasmic tail, associates with various intracellular proteins,
including with vari (10). Loss of
E-cadherin expression is typically observed in carcinomas (11), and in breast cancer, transfection
with ectopic E-cadherin has been demonstrated to decrease the
invasiveness of cancer cells (12).
Epidermal growth factor receptor (EGFR) is a member
of the receptor tyrosine kinase family, and overexpression of EGFR
has been documented in OSCC (13).
Stimulation of EGFR induces intrinsic tyrosine kinase activity and
cellular signaling, resulting in cell growth and proliferation
(14). EGFR stimulation is also
associated with perturbation of E-cadherin-mediated cell adhesion,
morphological fibroblast-like changes and increased cell motility
in tumors (15), due to association
of EGFR with the cadherin-catenin complex (16).
Targeting of EGFR signaling is a potential therapy
for the treatment of many cancers, including non-small-cell lung
cancer and colorectal cancer (17,18).
Specific drugs, such as erlotinib and gefitinib, reversibly inhibit
the EGFR tyrosine kinase domain by competitively binding with
adenosine triphosphate, while monoclonal antibodies, such as
cetuximab and panitumumab, block ligand binding to the
extracellular domain of EGFR and promote receptor internalization
(18). In recurrent or metastatic
head and neck SCCs, it has been observed that cetuximab in
combination with chemotherapy improved overall survival (19). Cetuximab combined with high-dose
radiotherapy has also been demonstrated to improve locoregional
control in locoregionally advanced head and neck SCCs (20). Despite the benefits of EGFR-targeting
agents, a minority of patients with head and neck cancer are
unresponsive to EGFR targeting therapies. Therefore, studies are
required to elucidate the underlying molecular mechanisms regarding
the effects of EGFR inhibition in cancer cells. The present study
aimed to evaluate the effect of EGFR inhibition on OSCC cells,
particularly on cell-cell junctions mediated by cadherin, by
performing wound healing, E-cadherin immunostaining and
transepithelial resistance assays in OSCC cells treated with EGFR
inhibitor (AG1478) or EGFR small interfering RNA (siRNA). The
efficacy and limitations of EGFR-targeted therapies were also
evaluated.
Materials and methods
Cell culture
The human HSC-3 OSCC cell line was obtained from the
National Institute of Biomedical Innovation (Osaka, Japan). HSC-3
cells were cultured in high glucose Dulbecco's modified Eagle's
medium (DMEM) supplemented with L-glutamine and Phenol Red (Wako
Pure Chemical Industries, Ltd., Osaka, Japan), 10% fetal bovine
serum and 1% penicillin/streptomycin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at 37°C in a humidified atmosphere of 5%
CO2. Cells were incubated in DMEM for 24–48 h prior to
treatment. Early passages of cells (between 2 and 10 passages from
the stage of primary culture) were used in the current study.
Reagents and antibodies
Rat anti-E-cadherin monoclonal antibody (ECCD2) was
purchased from Takara Bio, Inc. (Otsu, Japan) and the EGFR
inhibitor AG1478 was obtained from Merck KGaA. Mouse anti-zonula
occludens (ZO)-1 monoclonal antibody (T8-754) was characterized as
described previously (21). Rabbit
anti-EGFR monoclonal antibody (D38B1) was purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Secondary antibodies
conjugated with Alexa Fluor® 488 donkey anti-rabbit IgG (H+L;
A11059) and Cy®3 donkey anti-mouse IgG (H+L; AP192C) were purchased
from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
RNA interference (RNAi)
experiments
The following Stealth RNAi™ small interfering
(si)RNA (Invitrogen; Thermo Fisher Scientific, Inc.) was used for
RNAi experiments: Human EGFR-EGFRHSS103116,
5′-CCUAUGCCUUAGCAGUCUUAUCUAA-3′. A Stealth RNAi™ siRNA negative
control (Invitrogen; Thermo Fisher Scientific, Inc.) was used for
control experiments. Transfection of HSC-3 cells with the siRNAs
was performed using a Lipofectamine® RNAi MAX reagent according to
the manufacturer's protocol. (Invitrogen; Thermo Fisher Scientific,
Inc.).
Western blot analysis
Cells were lysed with lysis buffer containing 0.5 M
Tris-HCl (pH 6.8), 10% SDS and glycerol, and 1 M dithiothreitol was
added to cell lysates prior to loading at room temperature.
siRNA-treated cells were lysed in the same way. A total of 20 µg
protein were loaded on 10% SDS-PAGE gels. Protein mobility was
assessed using Precision Plus Protein™ Dual Color Standards
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein bands were
transferred to Immnobilon-P polyvinylidene difluoride membranes
(Merck KGaA) and incubated with rabbit anti-EGFR monoclonal
antibody (D38B1; 1:1,000) and rabbit anti-GAPDH polyclonal antibody
(ab9485; 1:2,500; Abcam, Cambridge, UK) for 24 h at 4°C, followed
by incubation with corresponding horseradish peroxidase-conjugated
goat anti-rabbit IgG (H+L) secondary antibody (31460; Invitrogen;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature.
Immunostained bands were developed using an enhanced
chemiluminescence system (GE Healthcare Life Sciences, Chalfont,
UK). Blots were scanned with a LAS-3000 mini imaging system
(Fujifilm, Tokyo, Japan). GAPDH was used as an internal loading
control. The experiment was repeated three times.
Immunofluorescence staining and
microscopy
Cells were grown with DMEM on microcover glass slips
in 35×10 mm polystyrene tissue culture dishes at a density of
1×105 cells/ml for 24 h. The cells were fixed with 1%
formaldehyde in phosphate-buffered saline (PBS) for 10 min at room
temperature, then treated with 0.2% Triton X-100 in PBS for 5 min
at room temperature and washed with PBS. The cells were blocked
with 1% bovine serum albumin (Sigma-Aldrich; Merck KGaA) in PBS for
30 min at room temperature, then incubated with primary antibodies
(rat anti-E-cadherin monoclonal antibody, mouse anti-ZO-1
monoclonal antibody, and rabbit anti-EGFR monoclonal antibody, as
mentioned above) for 24 h at 4°C. All antibodies were diluted with
1% bovine serum albumin in PBS as mentioned above. The cells were
then rinsed three times with PBS and incubated with corresponding
secondary antibodies (1:500; Alexa Fluor® 488 and Cy3®) for 30 min
at room temperature. After rinsing with PBS, specimens were
embedded in FluorSave™ (Merck KGaA) and observed with an IX71
fluorescence microscope (Olympus Soft Imaging Solutions GmbH,
Münster, Germany). Images were captured using a combined ORCA-ER
cooled CCD camera (Hamamatsu Photonics K.K., Shizuoka, Japan). For
dose-dependent AG1478 experiments, immunofluorescence staining was
performed 24 h after the addition of AG1478 (0, 0.5, 2 and 50 µM)
to 80% confluent HSC-3 cells.
Quantification of cell number and
cell-cell junctions
Cells were immunostained following AG1478 treatment
(0, 0.5, 2, 10 and 50 µM) for 24 h, and the number of cells was
counted in five randomly selected, independent microscopic images
(magnification, ×20). For quantification of cell junctions, the
numbers of cell-cell borders involving co-expression of E-cadherin
and ZO-1 were counted in five randomly selected, independent
microscopic images (magnification, ×20). Results are representative
of five independent experiments.
Measurement of transepithelial
resistance
Aliquots containing 1×105
cells/cm2 were plated onto Transwell filters (12 mm in
diameter; six filters for each cell treatment group) and cultured
in serum-free DMEM for 3 days until a confluent layer was formed.
Transepithelial resistance (TER) measurements were performed with a
Millicell-ERS electrical resistance meter (Merck KGaA) (22) immediately prior to and following the
addition of AG1478 (0, 0.5 and 2 µM) at given time points (24, 48
and 72 h). Resistance (ΔTER) of the HSC-3 layers was calculated as
the mean resistance of control inserts (without cells, n≥4)
subtracted from the mean resistance of cells treated with various
concentrations of AG1478 at given time points (results were
representative of ≥4 experiments).
In vitro wound healing assay
Cells (0.3×106 cells/ml) were plated in
duplicate in 6-well plates, grown to 100% confluence, and incubated
with high glucose DMEM containing AG1478 (0, 0.5 and 2 µM). After
24 h, cell monolayers were scraped with a sterile 200-µl disposable
plastic pipette tip and washed with PBS. The process of wound
healing was observed by microscopy for 0, 12 and 24 h at 37°C after
wounding, and images were obtained using a BZ-X700 fluorescence
microscope (Keyence Corporation, Osaka, Japan). Magnifications used
were ×4 for analyzing the wound healing assay and ×20 in
fluorescence microscopy.
Statistical analysis
Statistical analyses were performed using R software
(R Development Core Team, 2011). The Kruskal-Wallis test with
Steel-Dwass multiple comparisons was performed to compare the three
groups. P<0.05 was considered to indicate a statistically
significant significance. Data were presented as mean ± standard
deviation.
Results
Determination of the optimal
concentration of AG1478
Previous studies using specific EGFR inhibitors have
used relatively high concentrations of EGFR inhibitor. For
instance, oral and pancreatic cancer cell lines have previously
been treated with 20 (14) and 10 µM
AG1478 (23) respectively. However,
treatment with high concentrations of EGFR inhibitor may be
considered inappropriate for detailed observations of cell
junctions, due to the induction of cytotoxic effects. Previous
studies have demonstrated that AG1478 (0–32 µM) inhibits cell
growth in a dose-dependent manner, with lower concentrations of
AG1478 (8 µM) having little inhibitory effect on cell growth
(24). A study using serial
concentrations of AG1478 (0–40 µM) to treat human breast cancer
cells treated with for 72 h also documented that 20 µh AG1478 did
not induce significant apoptosis, relative to control cells, while
40 µM AG1478 induced significant apoptosis (25). It was also observed that few cells
survived at the higher concentration of AG1478 (40 µM) (25). To determine the optimal concentration
of AG1478 for use in the current study, the growth of HSC-3 cells
following treatment with AG1478 (0, 0.5, 2, 10 and 50 µM) for 24 h
was assessed by cell counting. Analogous to previous reports
(24), cytotoxic effects were
observed in HSC-3 cells following treatment with 50 µM AG1478,
while lower concentrations of AG1478 (0–10 µM) had little
inhibitory effect on cell growth (data not shown).
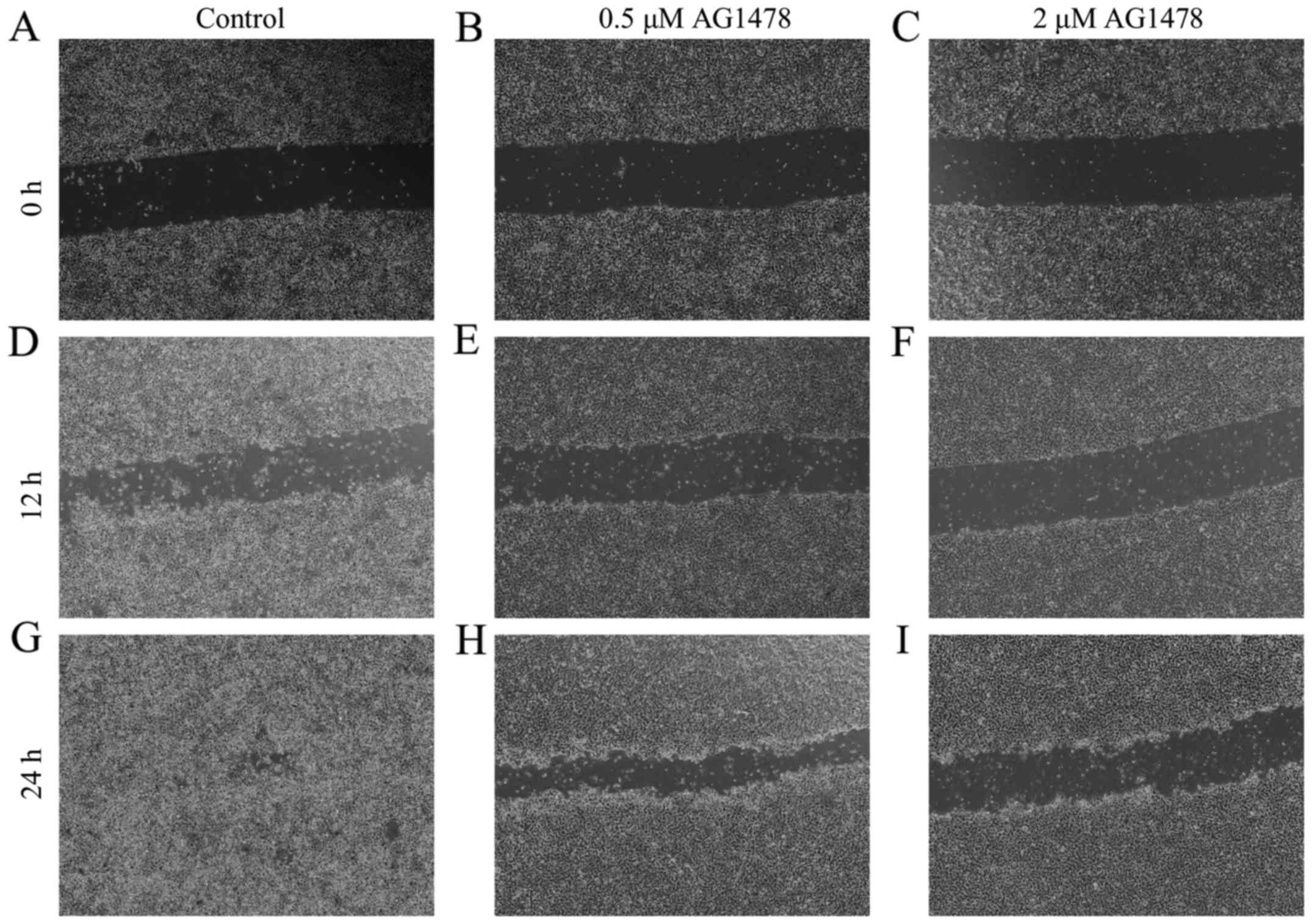
AG1478 suppresses cell motility
The integrated biological responses to EGFR
signaling are pleiotropic, resulting in tumor-promoting cellular
activities, including enhancement of cell motility and cytoskeletal
changes (26). Therefore, the
current study evaluated the motility of HSC-3 cells following
treatment with AG1478 (0.5 and 2 µM) for 12 and 24 h. In
vitro wound healing assays indicated that AG1478 treatment (2
µM) suppressed the motility of the OSCC cell line, relative to
untreated control cells (Fig.
1).
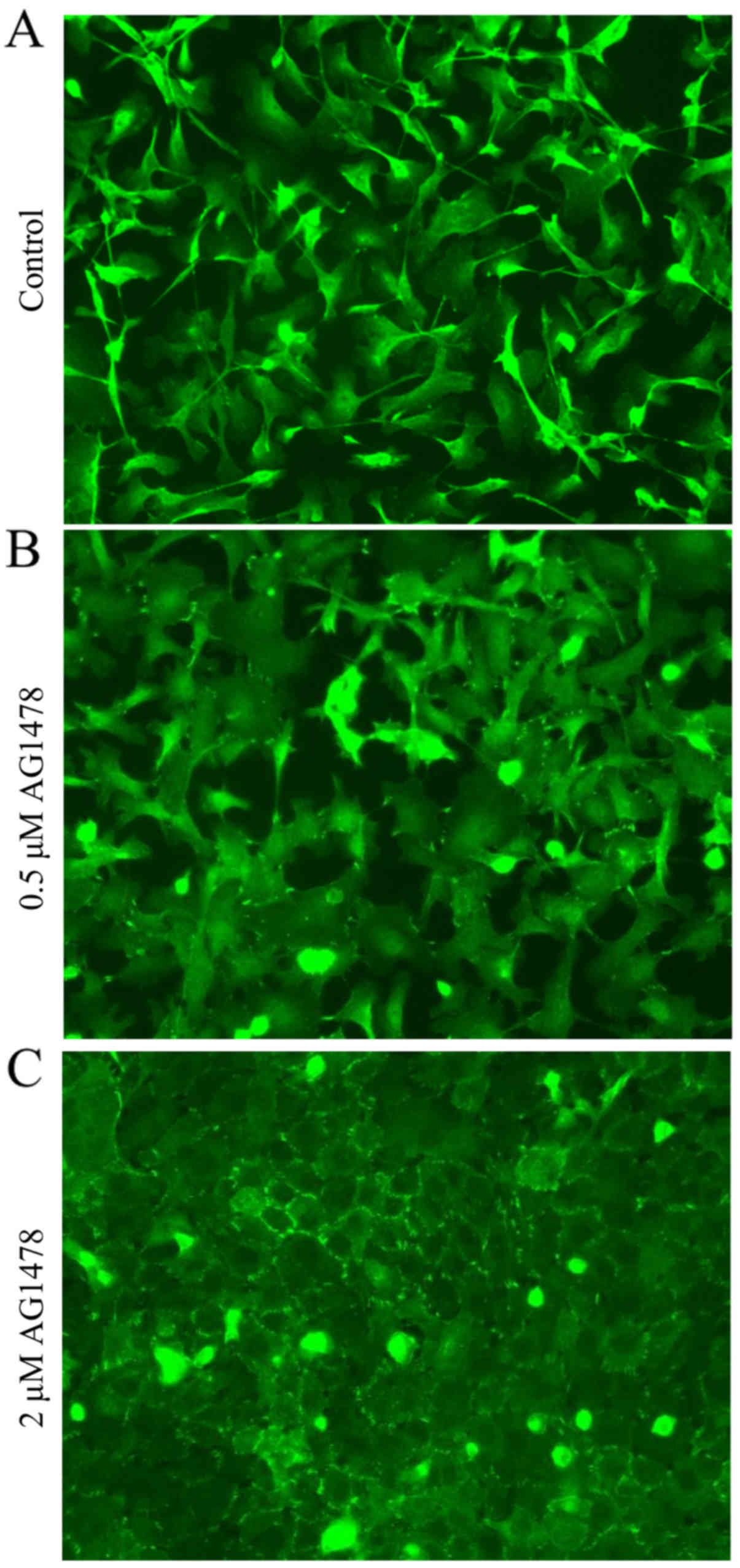
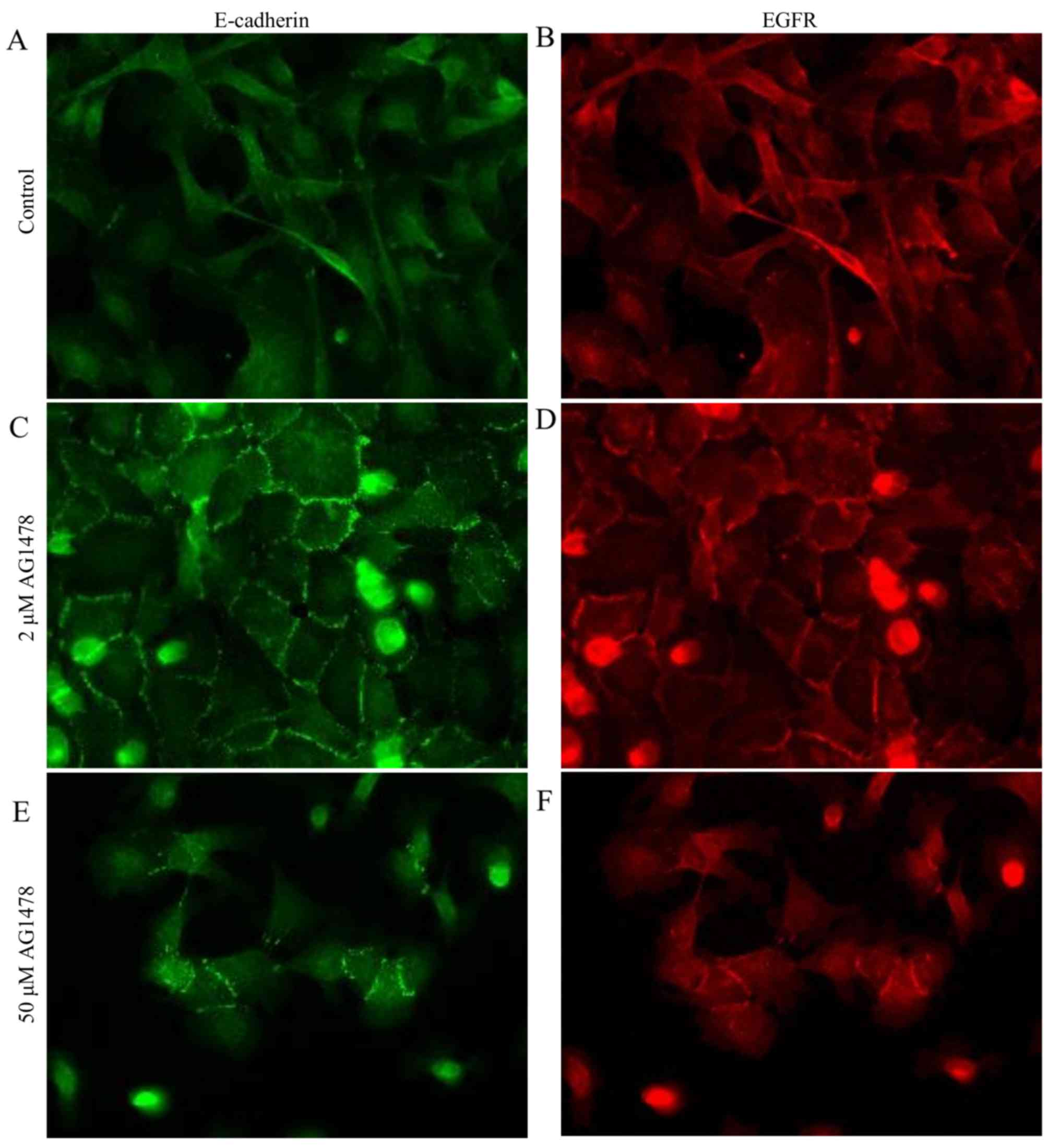
Morphological changes of HSC-3 cells
following AG1478 treatment
The expression pattern of E-cadherin in HSC-3 cells
treated with AG1478 (0.5 and 2 µM) was subsequently determined. It
was observed that AG1478 treatment altered the cellular morphology
of HSC-3 cells in a dose-dependent manner (Fig. 2). Control HSC-3 cells exhibited a
spindle-shaped fibroblastic cellular morphology, and prominent
spaces were observed between cells (Fig.
2A). Treatment of cells with 0.5 µM AG1478 flattened the
fibroblastic morphology of HSC-3 cells (Fig. 2B), and the higher concentration of
AG1478 (2 µM) caused cells to adopt an epithelial-like squamous
morphology (Fig. 2C). Relative to
all other concentrations of AG1478 investigated (0–50 µM), 2 µM
AG1478 reduced the spaces between cells to the greatest extent.
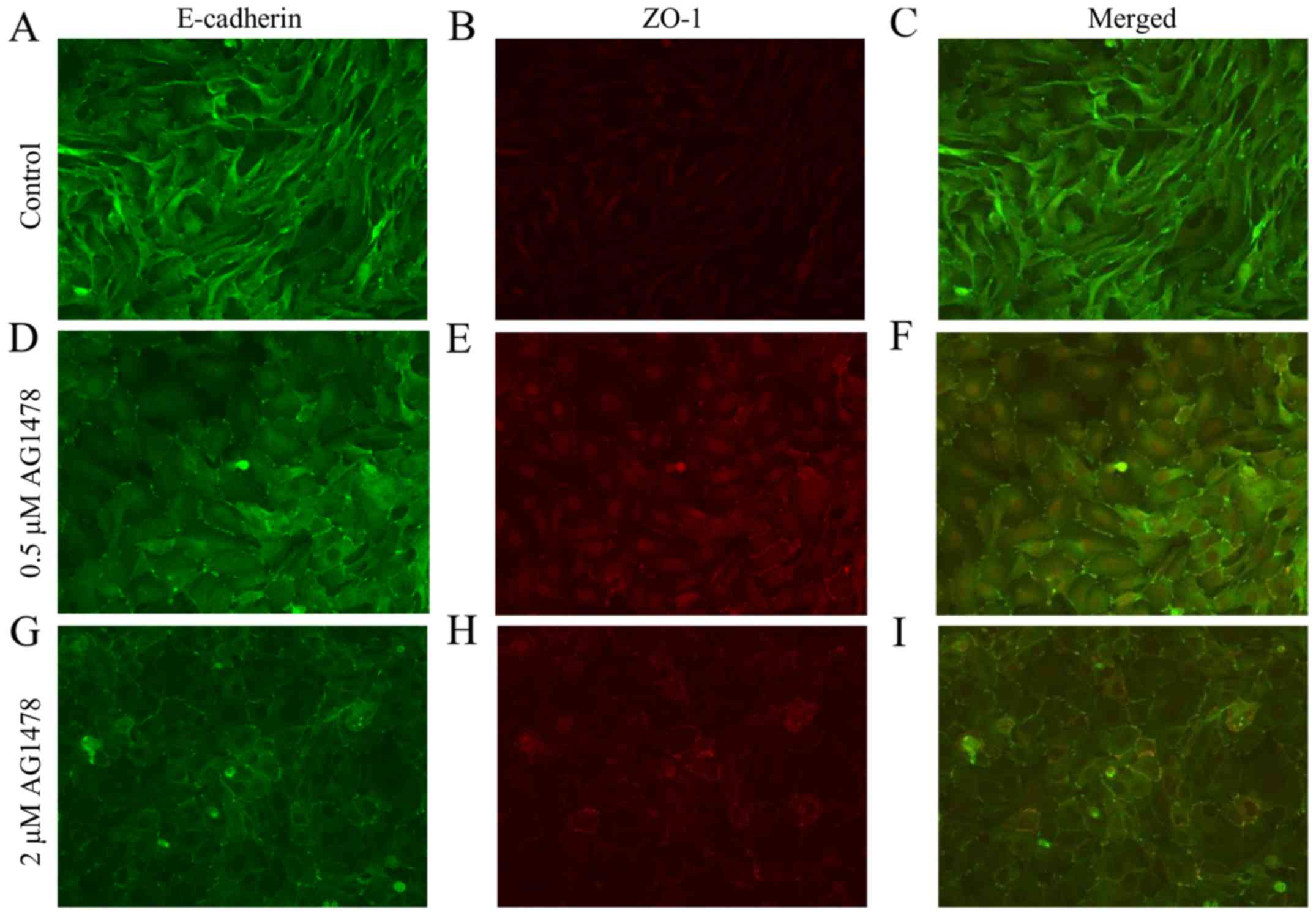
Immunostaining of cell-cell contacts demonstrated that AG1478
altered the expression of E-cadherin and the tight
junction-associated cytoplasmic protein ZO-1, as a marker of cell
junctions in various cell types (27), in a dose-dependent manner. In control
HSC-3 cells, E-cadherin and ZO-1 were not consistently colocalized,
due to the absence of ZO-1 and E-cadherin accumulations at the cell
periphery and cell-cell contacts, respectively (Fig. 3A). Treatment of cells with AG1478
(0.5 µM) induced the formation of punctate cell-cell junctions,
indicated by discontinuous zig-zag accumulations of E-cadherin and
ZO-1 at cell-cell contacts (Fig.
3B). Treatment with the higher concentration of AG1478 (2 µM)
led to the formation of continuous linear junctions, indicated by
linear accumulations and co-expression of E-cadherin and ZO-1
(Fig. 3C), which appeared similar to
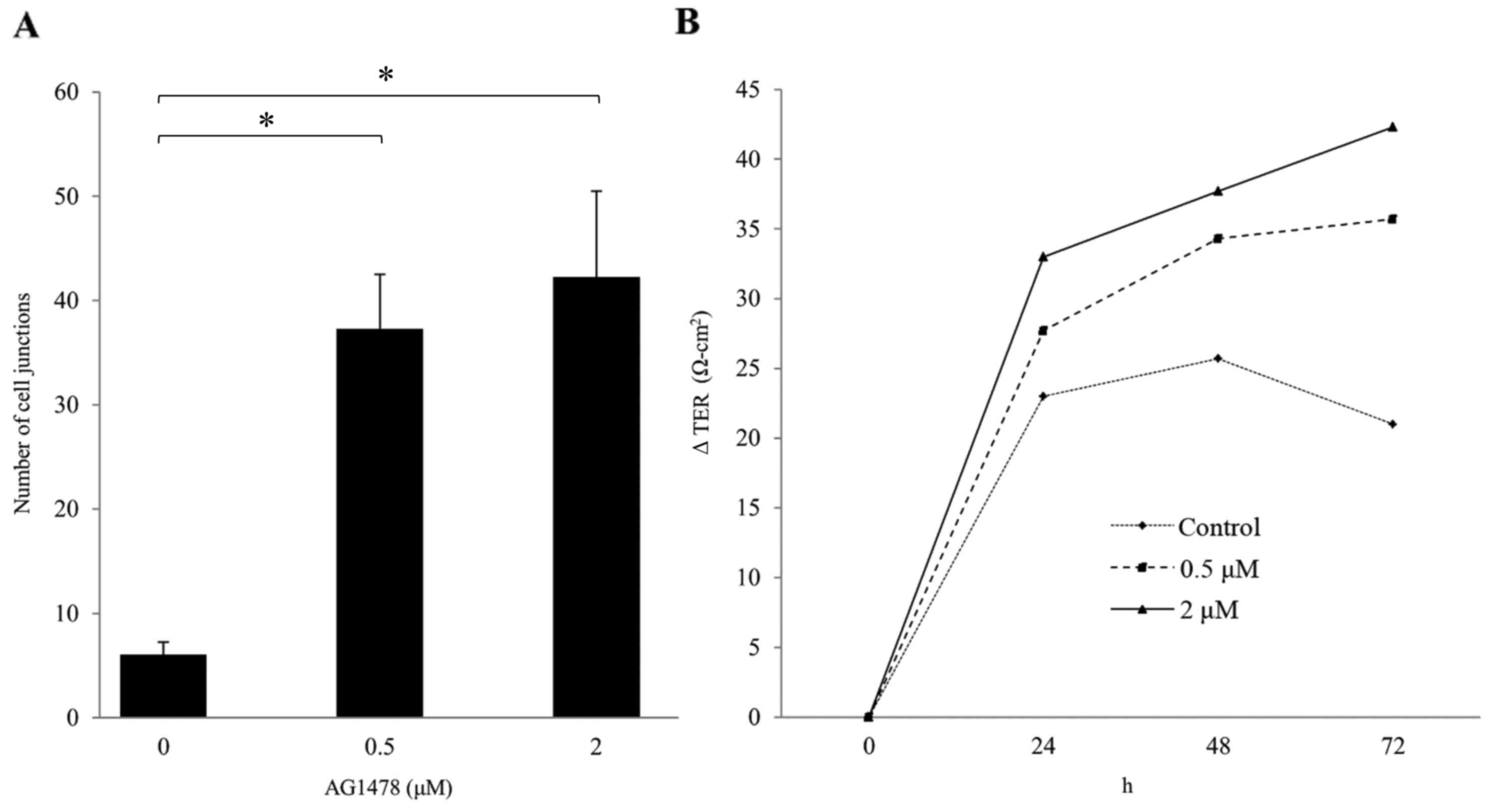
cell junctions in normal squamous epithelial cells. The number of
cell junctions (i.e., the numbers of cell-cell borders involving
co-expression of E-cadherin and ZO-1) significantly increased in a
dose-dependent manner (P<0.05; Fig.
4A).
AG1478 increases TER
TER was also investigated as an index of epithelial
barrier function. It was observed that AG1478 (0.5 and 2 µM)
increased TER in a dose-dependent manner (Fig. 4B), despite having no effect on total
cell number (data not shown).
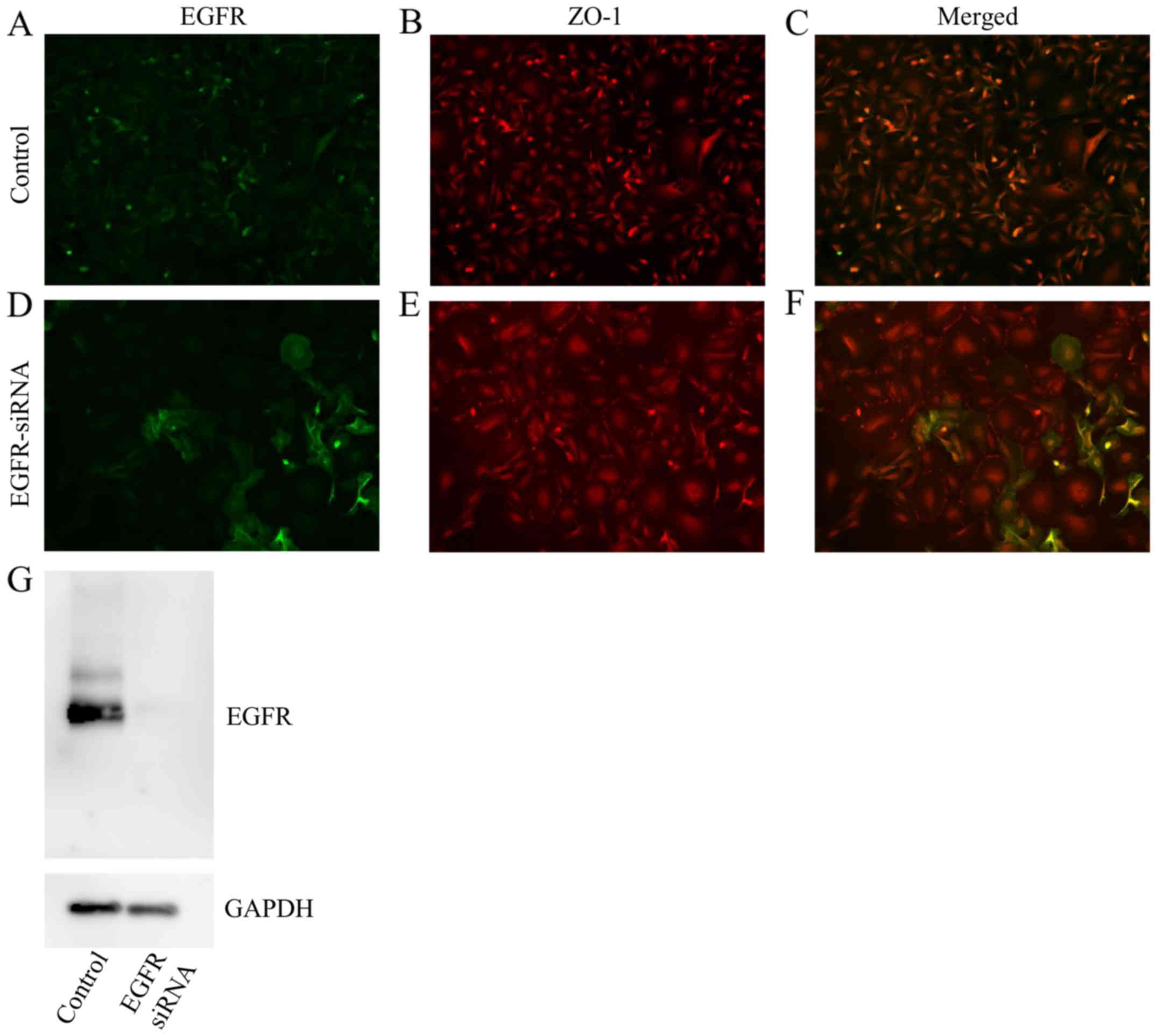
EGFR knockdown induces morphological
changes in HSC-3 cells
Similar to AG1478 treatment, knockdown of EGFR
flattened the fibroblastic morphology of HSC-3 cells (Fig. 5A-C), relative to untransfected
control cells (Fig. 5D-F),
indicating an epithelial-like squamous cell phenotype.
High-dose AG1478 treatment reduces the
number of HSC-3 cells
Finally, the phenotype of HSC-3 cells following
high-dose AG1478 (50 µM) treatment was assessed by immunostaining
(Fig. 6). High dose AG1478 caused a
marked reduction in the number of HSC-3 cells, relative to
untreated controls and cells treated with 2 µM AG1478. In addition,
E-cadherin accumulation and co-expression with EGFR at the
cell-cell boundaries was retained in a number of the surviving
cells. However, isolated cells lacking cell-cell adhesion were also
observed.
Discussion
Overexpression of EGFR has been documented in OSCC
(28,29). EGFR signaling is associated with a
loss of cell adhesion mediated by E-cadherin and acquisition of a
fibroblast-like cellular morphology, which subsequently increases
cell motility and potentially serves a role in tumor invasion and
metastasis (14,30,31). In
the present study, morphological changes in HSC-3 cells and
reductions in cell motility were observed following EGFR
inhibition. Treatment of cells with a low concentration of EGFR
inhibitor induced the formation of cell-cell junctions, indicated
by an accumulation of E-cadherin and ZO-1 at cell-cell contacts and
strengthening of barrier function. Suppression of EGFR expression
by siRNA also induced cellular morphological changes and
accumulation of E-cadherin at cell-cell contacts. The HSC-3 cells
used in the present study are generally poorly differentiated cells
with a random morphology, have exhibit a limited ability to form
cell junctions within monolayer cultures (32). Previous studies using HSC-3 cells
have documented low-level activation of EGFR within cells grown in
monolayer cultures, and an infrequent colocalization of
phosphorylated EGFR and E-cadherin (33). It has also been suggested that other
pathways may activate EGFR. For instance, in the process of cell
adhesion, integrins bind to extracellular matrix proteins, which in
turn may stimulate multiple pathways that modulate actin
cytoskeletal organization and cell motility (34). In response to cell-matrix adhesion, a
complex involving integrins and EGFR is formed, and EGFR is
subsequently phosphorylated on tyrosines 845, 1068, 1086 and 1173,
though not on 1148 (34).
Phosphorylation of EGFR at tyrosine 1173 has been associated with a
poor prognosis in OSCC (26). These
results indicate that treatment of HSC-3 cells with low
concentrations of EGFR inhibitor may affect integrin-mediated
signaling pathways and consequently alter cytoskeletal morphology
and cell motility.
It was also observed in the current study that HSC-3
cells with cell-cell junctions positive for E-cadherin were able to
survive high-dose EGFR inhibitor treatment. Previous reports have
demonstrated that inhibition of EGFR kinase activity with tyrosine
kinase inhibitors typically leads to decreased cell proliferation
without affecting cell survival (35), while targeted knockdown of the EGFR
protein has been found to result in cell death (36). Cell death induced by EGFR knockdown
may be due to autophagy and not typical apoptosis (37). Autophagy, a process of intracellular
proteolysis, is induced in various cancer cell lines, including
those for non-small-cell lung cancer and colorectal cancer,
following treatment with EGFR tyrosine kinase inhibitors in a
dose-dependent manner; however, autophagy is not activated in cells
that exhibit resistance to EGFR inhibitors (38). Inhibition of tyrosine kinase activity
alone has limited therapeutic efficacy, possibly due to the
inhibitory effects of EGFR on autophagy in various cancer cell
lines, which are potentially independent of its tyrosine kinase
activity (37). A previous study in
ovarian cancer cells observed that epidermal growth factor induced
a downregulation in E-cadherin expression and increased the
invasiveness of cancer cells (39).
In head and neck SCC cells overexpressing E-cadherin, it has also
been documented that a reduction in E-cadherin expression may lead
to an upregulation in EGFR transcription, suggesting that loss of
E-cadherin may induce proliferation by activating EGFR (40). Furthermore, in pancreatic carcinoma
cells, inhibitors of matrix metalloproteinases markedly reduced
E-cadherin expression while suppressing EGFR activation, while
upregulation of E-cadherin led to changes in cellular morphology,
decreased cell motility and enhanced apoptotic sensitivity in
response to chemotherapeutic treatment (41). In this previous study, upregulation
of E-cadherin was through suppression of ZEB1, as a transcriptional
repressor of E-cadherin (41).
Therefore, suppression of EGFR leading to increased expression of
E-cadherin at cell-cell junctions, as observed in the present
study, may be mediated by ZEB1.
E-cadherin is considered to be important during the
induction and progression of epithelial cancers (42). In addition, recovery of E-cadherin
expression in metastatic lesions arising from E-cadherin-deficient
primary tumors has been documented (43,44),
indicating that a mesenchymal-to-epithelial transition may occur
during metastasis (45). Distant
metastases exhibit equivalent or elevated levels of E-cadherin
expression when compared to the primary tumors from which they
originated (46). In some tumors,
cadherins attenuate cell growth, while in others, they promote
growth and survival of cancer cells (33). Previous studies have demonstrated
that cell-cell adhesion may promote resistance to anticancer
therapies, including chemotherapy and radiotherapy (47–50).
Shen and Kramer (33) have suggested
that cell survival mediated by E-cadherin, by the overexpression of
EGFR at cell-cell adhesion sites, may render cancer cells resistant
to treatment. Cell adhesion mediated by E-cadherin induces
ligand-independent EGFR activation, which triggers the
mitogen-activated protein kinase (MAPK) pathway and results in the
inhibition of apoptosis (26,33). It
has been recently documented that MAPK may mediate apoptosis and
autophagy in response to various stimuli through a number of
downstream pathways, including p38 and c-Jun N-terminal kinase MAPK
pathways (51). However, the
underlying mechanisms regarding the resistance of cancer cells to
apoptosis and autophagy warrant further investigation.
A limitation of the present study was the use of
only one OSCC cell line. To validate findings, future studies with
more differentiated cells that express higher levels of E-cadherin,
such as Ca9-22 or KB cells, are required. In addition, the present
study only evaluated one strategy of EGFR suppression, through the
use of a specific EGFR inhibitor. Thus, future studies are
warranted to investigate the efficacy of anti-EGFR antibodies such
as panitumumab.
In conclusion, treatment of OSCC cells with low
concentrations of EGFR inhibitor led to the acquisition of
epithelial properties, as indicated by E-cadherin-mediated cell
junctions, suppression of cell motility and an increase in TER. In
addition, cells that survived high-dose EGFR inhibitor treatment
retained high expression of E-cadherin at cell-cell boundaries.
This resistance was not observed in untreated OSCC cells. Future
studies into the properties of resistant cancer cells that retain
E-cadherin junctions may aid to identify cancer cell survival
mechanisms and treatment strategies that overcome resistance to
EGFR-targeting therapies.
Acknowledgements
The authors are thankful to Dr M. Itoh for supplying
the T8-754 antibody. The present study was supported by the Japan
Society for the Promotion of Science, Grants-in-Aid for Young
Scientists (grant no. 23792340).
References
|
1
|
Molinolo AA, Amornphimoltham P, Squarize
CH, Castilho RM, Patel V and Gutkind JS: Dysregulated molecular
networks in head and neck carcinogenesis. Oral Oncol. 45:324–334.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sales KU, Giudice FS, Castilho RM, Salles
FT, Squarize CH, Abrahao AC and Pinto DS Jr: Cyclin D1-induced
proliferation is independent of beta-catenin in head and neck
cancer. Oral Dis. 20:e42–e48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nagpal JK and Das BR: Oral cancer:
Reviewing the present understanding of its molecular mechanism and
exploring the future directions for its effective management. Oral
Oncol. 39:213–221. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka T, Iino M and Goto K: Knockdown of
Sec 6 improves cell-cell adhesion by increasing α-5-catenin in oral
cancer cells. FEBS Lett. 586:924–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chaw SY, Majeed AA, Dalley AJ, Chan A,
Stein S and Farah CS: Epithelial to mesenchymal transition (EMT)
biomarkers-E-cadherin, beta-catenin, APC and vimentin-in oral
squamous cell carcinogenesis and transformation. Oral Oncol.
48:997–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakamoto K, Imanishi Y, Tomita T, Shimoda
M, Kameyama K, Shibata K, Sakai N, Ozawa H, Shigetomi S, Fujii R,
et al: Overexpression of SIP1 and downregulation of E-cadherin
predict delayed neck metastasis in stage I/II oral tongue squamous
cell carcinoma after partial glossectomy. Ann Surg Oncol.
19:612–619. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Munshi HG, Ghosh S, Mukhopadhyay S, Wu YI,
Sen R, Green KJ and Stack MS: Proteinase suppression by
E-cadherin-mediated cell-cell attachment in premalignant oral
keratinocytes. J Biol Chem. 277:38159–38167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nagafuchi A: Molecular architecture of
adherens junctions. Curr Opin Cell Biol. 13:600–603. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Aken E, De Wever O, da Rocha AS
Correia and Mareel M: Defective E-cadherin/catenin complexes in
human cancer. Virchows Arch. 439:725–751. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frixen UH and Nagamine Y: Stimulation of
urokinase-type plasminogen activator expression by blockage of
E-cadherin-dependent cell-cell adhesion. Cancer Res. 53:3618–3623.
1993.PubMed/NCBI
|
|
13
|
Forastiere AA and Burtness BA: Epidermal
growth factor receptor inhibition in head and neck cancer-more
insights, but more questions. J Clin Oncol. 25:2152–2155. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee CH, Hung HW, Hung PH and Shieh YS:
Epidermal growth factor receptor regulates beta-catenin location,
stability, and transcriptional activity in oral cancer. Mol Cancer.
9:642010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lilien J and Balsamo J: The regulation of
cadherin-mediated adhesion by tyrosine
phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell
Biol. 17:459–465. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoschuetzky H, Aberle H and Kemler R:
Beta-catenin mediates the interaction of the cadherin-catenin
complex with epidermal growth factor receptor. J Cell Biol.
127:1375–1380. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Itoh M, Yonemura S, Nagafuchi A and
Tsukita S and Tsukita S: A 220-kD undercoat-constitutive protein:
Its specific localization at cadherin-based cell-cell adhesion
sites. J Cell Biol. 115:1449–1462. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Higashi T, Tokuda S, Kitajiri S, Masuda S,
Nakamura H, Oda Y and Furuse M: Analysis of the ‘angulin’ proteins
LSR, ILDR1 and ILDR2-tricellulin recruitment, epithelial barrier
function and implication in deafness pathogenesis. J Cell Sci.
126:966–977. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takai E, Tan X, Tamori Y, Hirota M, Egami
H and Ogawa M: Correlation of translocation of tight junction
protein Zonula occludens-1 and activation of epidermal growth
factor receptor in the regulation of invasion of pancreatic cancer
cells. Int J Oncol. 27:645–651. 2005.PubMed/NCBI
|
|
24
|
Takaoka S, Iwase M, Uchida M, Yoshiba S,
Kondo G, Watanabe H, Ohashi M, Nagumo M and Shintani S: Effect of
combining epidermal growth factor receptor inhibitors and cisplatin
on proliferation and apoptosis of oral squamous cell carcinoma
cells. Int J Oncol. 30:1469–1476. 2007.PubMed/NCBI
|
|
25
|
Zhang YG, Du Q, Fang WG, Jin ML and Tian
XX: Tyrphostin AG1478 suppresses proliferation and invasion of
human breast cancer cells. Int J Oncol. 33:595–602. 2008.PubMed/NCBI
|
|
26
|
Monteiro L, Ricardo S, Delgado M, Garcez
F, do Amaral B and Lopes C: Phosphorylated EGFR at tyrosine 1173
correlates with poor prognosis in oral squamous cell carcinomas.
Oral Dis. 20:178–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kakei Y, Akashi M, Shigeta T, Hasegawa T
and Komori T: Alteration of cell-cell junctions in cultured human
lymphatic endothelial cells with inflammatory cytokine stimulation.
Lymphat Res Biol. 12:136–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Störkel S, Reichert T, Reiffen KA and
Wagner W: EGFR and PCNA expression in oral squamous cell
carcinomas-A valuable tool in estimating the patient's prognosis.
Eur J Cancer B Oral Oncol. 29B:1–277. 1993.PubMed/NCBI
|
|
29
|
Monteiro LS, Diniz-Freitas M,
Garcia-Caballero T, Forteza J and Fraga M: EGFR and Ki-67
expression in oral squamous cell carcinoma using tissue microarray
technology. J Oral Pathol Med. 39:571–578. 2010.PubMed/NCBI
|
|
30
|
Hamaguchi M, Matsuyoshi N, Ohnishi Y,
Gotoh B, Takeichi M and Nagai Y: p60v-src causes tyrosine
phosphorylation and inactivation of the N-cadherin-catenin cell
adhesion system. EMBO J. 12:307–314. 1993.PubMed/NCBI
|
|
31
|
Shibata T, Gotoh M, Ochiai A and Hirohashi
S: Association of plakoglobin with APC, a tumor suppressor gene
product and its regulation by tyrosine phosphorylation. Biochem
Biophys Res Commun. 203:519–522. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawano K, Kantak SS, Murai M, Yao CC and
Kramer RH: Integrin alpha3beta1 engagement disrupts intercellular
adhesion. Exp Cell Res. 262:180–196. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen X and Kramer RH: Adhesion-mediated
squamous cell carcinoma survival through ligand-independent
activation of epidermal growth factor receptor. Am J Pathol.
165:1315–1329. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moro L, Dolce L, Cabodi S, Bergatto E,
Erba E Boeri, Smeriglio M, Turco E, Retta SF, Giuffrida MG,
Venturino M, et al: Integrin-induced epidermal growth factor (EGF)
receptor activation requires c-Src and p130Cas and leads to
phosphorylation of specific EGF receptor tyrosines. J Biol Chem.
277:9405–9414. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harari PM and Huang SM: Combining EGFR
inhibitors with radiation or chemotherapy: Will preclinical studies
predict clinical results? Int J Radiat Oncol Biol Phys. 58:976–983.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nagy P, Arndt-Jovin DJ and Jovin TM: Small
interfering RNAs suppress the expression of endogenous and
GFP-fused epidermal growth factor receptor (erbB1) and induce
apoptosis in erbB1-overexpressing cells. Exp Cell Res. 285:39–49.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH,
Fidler IJ and Hung MC: Survival of cancer cells is maintained by
EGFR independent of its kinase activity. Cancer Cell. 13:385–393.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fung C, Chen X, Grandis JR and Duvvuri U:
EGFR tyrosine kinase inhibition induces autophagy in cancer cells.
Cancer Biol Ther. 13:1417–1424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheng JC, Qiu X, Chang HM and Leung PC:
HER2 mediates epidermal growth factor-induced down-regulation of
E-cadherin in human ovarian cancer cells. Biochem Biophys Res
Commun. 434:81–86. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang D, Su L, Huang D, Zhang H, Shin DM
and Chen ZG: Downregulation of E-Cadherin enhances proliferation of
head and neck cancer through transcriptional regulation of EGFR.
Mol Cancer. 10:1162011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang F, Sloss C, Zhang X, Lee SW and
Cusack JC: Membrane-bound heparin-binding epidermal growth factor
like growth factor regulates E-cadherin expression in pancreatic
carcinoma cells. Cancer Res. 67:8486–8493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saha B, Chaiwun B, Imam SS, Tsao-Wei DD,
Groshen S, Naritoku WY and Imam SA: Overexpression of E-cadherin
protein in metastatic breast cancer cells in bone. Anticancer Res.
27:3903–3908. 2007.PubMed/NCBI
|
|
44
|
Chao YL, Shepard CR and Wells A: Breast
carcinoma cells re-express E-cadherin during mesenchymal to
epithelial reverting transition. Mol Cancer. 9:1792010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gunasinghe NP, Wells A, Thompson EW and
Hugo HJ: Mesenchymal-epithelial transition (MET) as a mechanism for
metastatic colonisation in breast cancer. Cancer Metastasis Rev.
31:469–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kowalski PJ, Rubin MA and Kleer CG:
E-cadherin expression in primary carcinomas of the breast and its
distant metastases. Breast Cancer Res. 5:R217–R222. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
St Croix B and Kerbel RS: Cell adhesion
and drug resistance in cancer. Curr Opin Oncol. 9:549–556. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
St Croix B, Sheehan C, Rak JW, Flørenes
VA, Slingerland JM and Kerbel RS: E-Cadherin-dependent growth
suppression is mediated by the cyclin-dependent kinase inhibitor
p27(KIP1). J Cell Biol. 142:557–571. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Green SK, Frankel A and Kerbel RS:
Adhesion-dependent multicellular drug resistance. Anticancer Drug
Des. 14:153–168. 1999.PubMed/NCBI
|
|
50
|
Damiano JS, Hazlehurst LA and Dalton WS:
Cell adhesion-mediated drug resistance (CAM-DR) protects the K562
chronic myelogenous leukemia cell line from apoptosis induced by
BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation.
Leukemia. 15:1232–1239. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|