Introduction
Rheumatic heart disease (RHD) is an autoimmune
inflammatory disease with multiorgan involvement (1). RHD is caused by an abnormal immune
response to group A streptococcal infection (2), which leads to valve damage,
particularly to the mitral valve, and hemodynamic changes (3). In China, RHD remains a significant
health burden, although the prevalence of RHD has been declining in
recent years (4). Valve surgery is
considered the primary clinical treatment for RHD (5). However, patients with RHD frequently
suffer from serious complications and irreversible valve
dysfunction due to a lack of early detection (3).
Pulmonary arterial hypertension (PAH) is the most
frequent clinical complication of RHD (6). Right ventricular hypertrophy and heart
failure during the later stages of PAH are attributable to
significant reductions in the cross sectional area of pulmonary
vasculature, which may eventually be fatal (7,8). PAH is
a complex disease; multiple factors contribute to its onset and
development, including pathological environmental factors, genetic
polymorphisms and epigenetic changes (9–11).
Recent studies have demonstrated the role of epigenetic
modifications in the pathogenesis of PAH (11–13),
suggesting that DNA methylation may be associated with the etiology
of RHD with secondary PAH (RHD-PAH).
DNA methylation typically occurs at the 5′ position
of the cytosine ring in 5′-C-phosphate-G-3′ (CpG) dinucleotides and
is often associated with the regulation of gene expression
(14). DNA methylation serves an
important role in the development and progression of rheumatic
diseases (15–17). However, studies investigating the
role of epigenetics in RHD-PAH are scarce. The present study aimed
to identify differentially methylated regions (DMRs) in patients
with RHD-PAH. The present study identified novel DNA methylation
markers and will aid in improving the current understanding of
RHD-PAH.
Materials and methods
Sample collection, DNA extraction and
bisulfite modification
The clinical diagnosis of RHD-PAH was performed as
previously described (3).
Specifically, the inclusion criteria were as follows: i) Diagnosis
of mitral valve prolapse and scheduled for mitral valve
replacement; ii) left ventricular ejection fraction volume >50%
and left ventricular end-diastolic diameter <55 mm; iii)
pulmonary artery systolic pressure >40 mmHg prior to surgery;
and iv) no history of cardiomyopathy, congenital heart disease,
liver disease or renal disease. The present study was approved by
the Ethics Committee of Lihuili Hospital and informed consent forms
were signed by all participants. The clinical and pathological
characteristics of the involved individuals are described in
Table I. Since acute rheumatic fever
occurs more frequently in females (2), a total of 6 female patients with
RHD-PAH (the study group; age, 57.00±8.39 years) and 6 normal
female donors (the control group; age, 55.00±6.39 years) were
recruited for the present study from Lihuili Hospital (Ningbo,
China) between March 2014 and September 2015.
 | Table I.Clinical data for patients with
rheumatic heart disease and secondary pulmonary arterial
hypertension and healthy controls. |
Table I.
Clinical data for patients with
rheumatic heart disease and secondary pulmonary arterial
hypertension and healthy controls.
| Subgroup | Age (years) | Gender | PASP before surgery
(mmHg) |
|---|
| Case 1 | 60 | Female | 42 |
| Case 2 | 56 | Female | 117 |
| Case 3 | 58 | Female | 72 |
| Case 4 | 56 | Female | 55 |
| Case 5 | 69 | Female | 50 |
| Case 6 | 43 | Female | 64 |
| Control 1 | 60 | Female | NA |
| Control 2 | 51 | Female | NA |
| Control 3 | 64 | Female | NA |
| Control 4 | 54 | Female | NA |
| Control 5 | 46 | Female | NA |
| Control 6 | 55 | Female | NA |
Blood samples from the two groups of patients were
collected into EDTA tubes. DNA extraction and quantification
procedures were subsequently performed as previously described
(18). Genomic DNA bisulfite
conversion (500 ng) was performed using an EZ DNA
Methylation-Gold™ kit (Zymo Research, Irvine, CA, USA),
according to the manufacturer's protocol.
Methylation assay
A methylation assay was performed using the Infinium
HumanMethylation450 BeadChip kit (Illumina, Inc., San Diego, CA,
USA) as described previously (19).
In brief, bisulfite-converted DNA (200 ng) was used in the
whole-genome amplification reaction, followed by an enzymatic
end-point fragmentation, precipitation and resuspension in
hybridization buffer (19).
Subsequently, all steps were performed following the standard
Infinium protocol. Sample labeling, hybridization to chips and
image scanning were also carried out according to the Infinium
kit's protocol (20). All samples
were processed on a single chip to avoid the batch effect. A total
of 485,577 methylation loci, covering 21,231 genes, were tested
using this kit. Gene methylation datasets were submitted to the
Gene Expression Omnibus (accession number GSE84003).
Statistical analysis
Statistical analyses were conducted using R software
version 3.0.1 (www.r-project.org). Methylation levels of CpG sites
are expressed as β values ranging from 0–1. Methylation levels were
measured using β-values, which are based on the fluorescence
intensity of methylated and unmethy-lated probes. The calculation
of β values was performed as previously described (21). Prior to further calculations, probes
designed for sequences on sex chromosomes, with random single
nucleotide polymorphisms in the probe-mapping genomic regions or
with detection values of P>0.01 on all arrays were removed. The
remaining 463,289 probes were used for subsequent analysis.
Methylation levels were then compared with the differential
detection procedure in the Limma package version 3.30.13
(www.bioconductor.org/packages/release/bioc/html/limma.html)
using R software, as previously described (21). P<0.05 and an absolute value of
∆β>0.2 were considered to indicate a statistically significant
difference. The selection process is presented in Fig. 1. IlluminaHumanMethylation450k.db
annotation package version 2.0.9 (http://www.bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylation450k.db.html)
was used to provide detailed information about the 450k chip
platform, including mappings between gene symbol identifiers and
manufacturer identifiers and genomic positions. All annotations are
based on human genome build 19. A heatmap was created via heatmap.2
function in the gplots package. Signaling pathway and gene ontology
(GO) enrichment analyses were conducted using the Database for
Annotation, Visualization and Integrated Discovery (DAVID; version
6.7; http://david.abcc.ncifcrf.gov) and
the Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.jp/kegg), respectively.
Results
Quality control of methylation
data
Genome-wide DNA methylation profiles of the 12
samples were generated using the Infinium HumanMethylation450
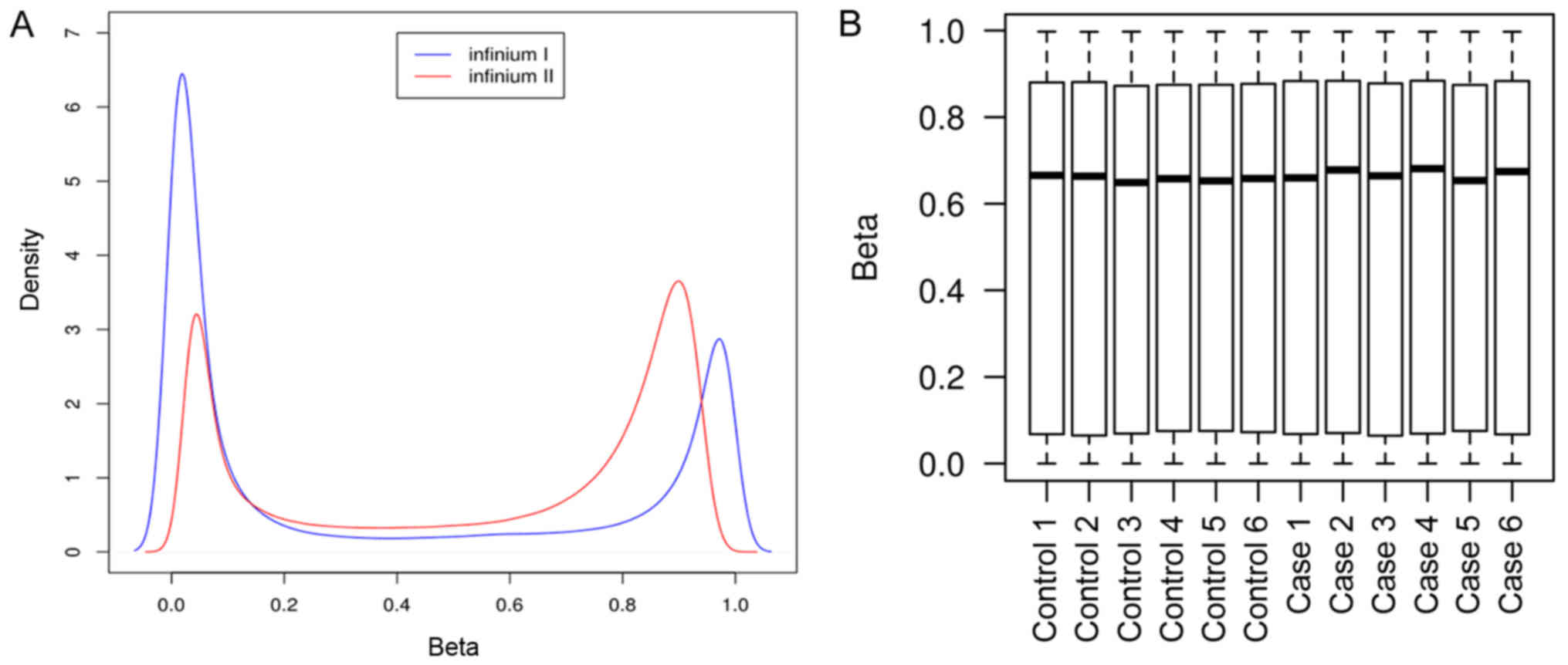
BeadChip kit (Fig. 1). The density
distribution of the β-values is presented in Fig. 2A. The results revealed a typical
bimodal-shape distribution of β-values. Furthermore, boxplots of
β-value distributions demonstrated homogenous levels of methylation
across all of the samples (Fig.
2B).
DMRs between patients with RHD-PAH and
healthy controls
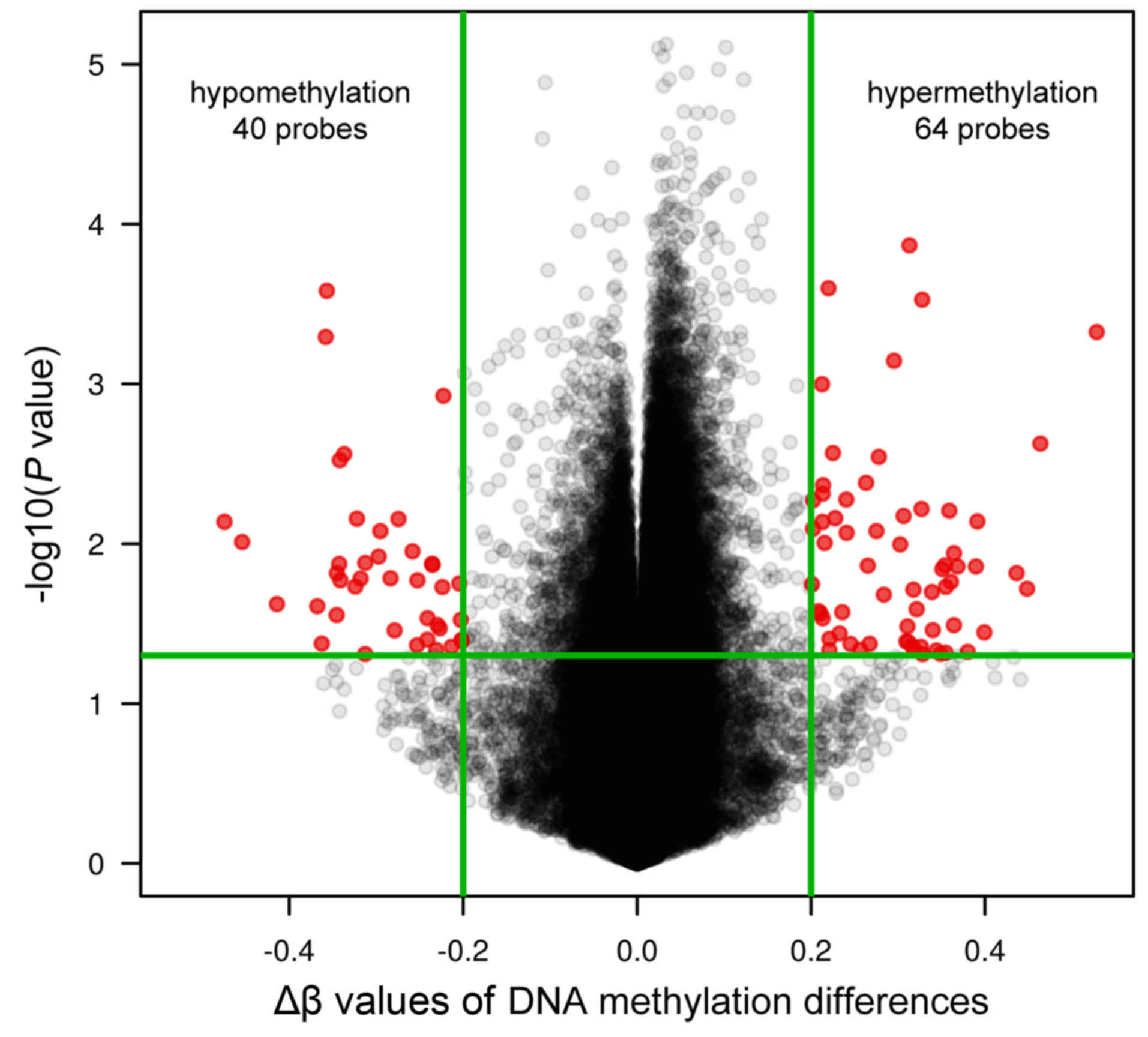
The graph in Fig. 3
represents the distribution of CpG sites sorted by mean ∆β values
and P-values (Fig. 3). In the
RHD-PAH group there were 40 hypomethylated and 64 hypermethylated
CpG sites compared with the healthy controls. These 104 identified
DMRs were mapped to 60 known genes (data not shown), such as
protein kinase C alpha (PRKCA), fibroblast growth factor receptor
2, protamine 1, S-phase kinase associated protein 2 and
hyperpolarization activated cyclic nucleotide gated potassium
channel 2. A total of 17 (16.35%) of the DMRs were in promoter
regions (5′UTR, TSS200, TSS1500 and 1stExon), 41 (39.42%) in gene
body regions, 5 (4.81%) in 3′UTR regions and 41 (39.42%) were
unknown.
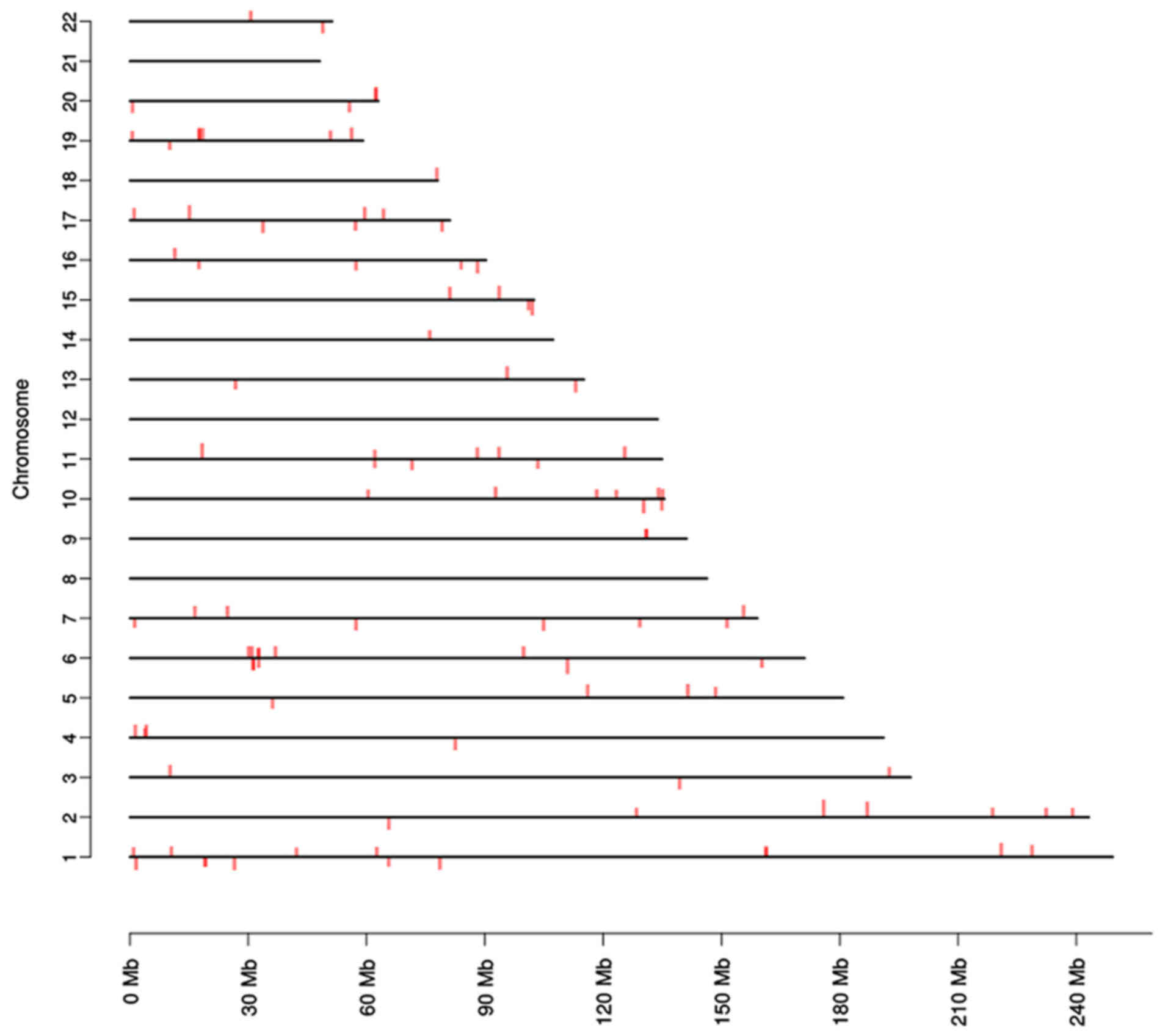
The chromosomal distribution of DMRs was evaluated
according to the annotation of genomic positions. It was revealed
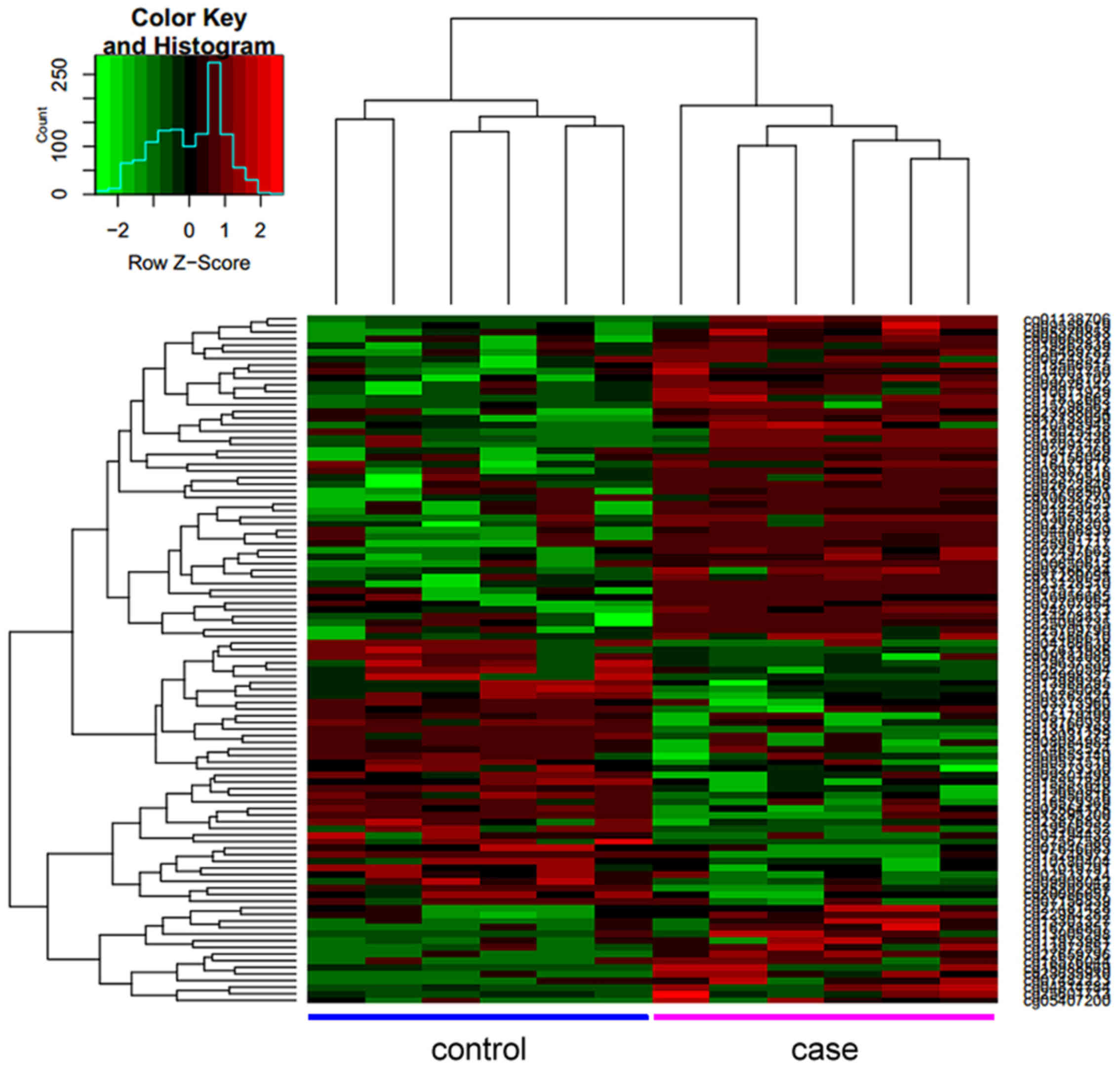
to affect a large range of genes and chromosomal regions (Fig. 4). Patient samples were also ordered
by hierarchical clustering, and a heat map was produced to allow
the visualization of the hypermethylated DMRs associated with
RHD-PAH (Fig. 5). The DMRs were
mapped to a number of genes, including PRKCA, protein kinase
AMP-activated non-catalytic subunit γ 2 (PRKAG2), sprouty related
EVH1 domain containing 2 (SPRED2) and LIF interleukin 6 family
cytokine (LIF).
Enrichment analysis of the genes with
DMRs
DAVID GO enrichment analysis of the genes that the
DMRs were mapped to revealed several significantly enriched GO
terms, including negative regulation of protein kinase activity,
positive regulation of protein amino acid phosphorylation, negative
regulation of transferase activity and positive regulation of
phosphate metabolic process (P<0.05; Table II). PRKCA, PRKAG2, SPRED2 and LIF
were each enriched in at least two of these processes. No
significantly enriched signaling pathways were identified by
KEGG.
| Table II.GO term enrichment analysis of the
differentially methylated regions in patients with rheumatic heart
disease and secondary pulmonary arterial hypertension compared with
the healthy controls. |
Table II.
GO term enrichment analysis of the
differentially methylated regions in patients with rheumatic heart
disease and secondary pulmonary arterial hypertension compared with
the healthy controls.
| Enriched term | Description | P-value | Genes involved |
|---|
| GO:0006469 | Negative regulation
of protein kinase activity | 0.031 | PRKCA, PRKAG2,
SPRED2 |
| GO:0001934 | Positive regulation
of protein amino acid phosphorylation | 0.032 | PRKCA, LIF,
PRKAG2 |
| GO:0051348 | Negative regulation
of transferase activity | 0.037 | PRKCA, PRKAG2,
SPRED2 |
| GO:0045937 | Positive regulation
of phosphate metabolic process | 0.040 | PRKCA, LIF,
PRKAG2 |
Discussion
In the present study, a genome-wide high-throughput
assay was used to identify DMRs in Chinese patients with RHD-PAH.
Rheumatic diseases, including RHD, frequently possess an element of
autoimmunity (22). The
hypomethylation of immune response-associated genes (cluster of
differentiation 9, matrix metallopeptidase 9, platelet derived
growth factor receptor α and bone marrow stromal cell antigen 2)
has previously been identified in systemic lupus erythematosus
(23), and DMRs in osteoarthritis
are primarily associated with inflammatory/defensive immune
responses (24). It has also been
reported that the altered DNA methylation of genes, including
interleukin (IL) 6 receptor, calpain 8, dipeptidyl peptidase 4 and
multiple homeobox genes, may mediate the risk of rheumatoid
arthritis (25).
In the present study, four candidate genes (PRKCA,
LIF, PRKAG2 and SPRED2) were identified to be associated with the
pathogenesis of RHD. Differentially methylated PRKCA contributes to
the risk of developing fibromyalgia in women (26). Patients with fibromyalgia typically
suffer from rheumatic symptoms, which are likely to be associated
with inflammatory cytokines (27).
LIF, which encodes a member of the IL-6 cytokine family,
downregulates autoimmune responses by enhancing the number of
regulatory T cells (28). This
suggests that the silencing of LIF by hypermethylation may increase
the risk of RHD. PRKAG2 mutation is responsible for glycogen
storage disease of the heart (29,30).
However, the role of PRKAG2 in the development RHD-PAH remains
unclear. SPRED2 has been reported to be a repressor of immune
responses (31). The SPRED2 rs934734
polymorphism was identified to be significantly associated with an
increased risk of rheumatoid arthritis (32), which suggests that SPRED2 may serve a
role in the pathogenesis of RHD. However, further studies are
required to explore the underlying mechanisms of these DMRs in the
pathogenesis of RHD-PAH.
The present study had several limitations. The
results of the current study were based on a genome-wide
methylation array of 6 patients with RHD-PAH and 6 healthy
controls, and large population validation for clinical application
should be performed. Furthermore, the present study only included
female Chinese patients with RHD-PAH, and so the DMRs identified
here require confirmation in males and patients of different
ethnicities. Finally, although DNA methylation regulation is an
important mechanism in the pathogenesis of RHD-PAH, the involvement
of other epigenetic regulation, including histone modification and
the effects of miRNA, remains to be explored.
In conclusion, the results of the present study
identified 40 hypomethylated and 64 hypermethylated CpG sites
between the RHD-PAH group and the control group. These DMRs may
provide novel insights into the pathogenesis of RHD-PAH.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81371469), Zhejiang
Provincial Natural Science Foundation (grant no. LY14H160008),
Ningbo City Medical Science and Technology Projects (grant no.
2014A20), the Advanced Key Scientific and Technological Programs of
Ningbo (grant no. 2012C5017), the Natural Science Foundation of
Ningbo (grant no. 2014A610272), the Natural Science Foundation of
Ningbo (grant no. 2016A610197), the Science and Technology
Foundation of Ningbo (grant no. 2016C51012) and the Science and
Technology Innovation Team of Ningbo (grant no. 2011B82015).
References
|
1
|
Villa-Forte A and Mandell BF:
Cardiovascular disorders and rheumatic disease. Rev Esp Cardiol.
64:809–817. 2011.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marijon E, Mirabel M, Celermajer DS and
Jouven X: Rheumatic heart disease. Lancet. 379:953–964. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li N, Lian J, Zhao S, Zheng D, Yang X,
Huang X, Shi X, Sun L, Zhou Q, Shi H, et al: Detection of
differentially expressed micrornas in rheumatic heart disease:
miR-1183 and miR-1299 as potential diagnostic biomarkers. Biomed
Res Int. 2015:5245192015.PubMed/NCBI
|
|
4
|
Lu H, Pan WZ, Wan Q, Cheng LL, Shu XH, Pan
CZ, Qian JY and Ge JB: Trends in the prevalence of heart diseases
over a ten-year period from single-center observations based on a
large echocardiographic database. J Zhejiang Univ Sci B. 17:54–59.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Victor S: Dilemmas in the management of
rheumatic heart disease. J Indian Med Assoc. 97:265–270.
1999.PubMed/NCBI
|
|
6
|
Sriharibabu M, Himabindu Y and Kabir Z:
Rheumatic heart disease in rural south India: A
clinico-observational study. J Cardiovasc Dis Res. 4:25–29. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Izikki M, Guignabert C, Fadel E, Humbert
M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B,
et al: Endothelial-derived FGF2 contributes to the progression of
pulmonary hypertension in humans and rodents. J Clin Invest.
119:512–523. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu
L, Chu L, Zamel R, Machuca T, Waddell T, et al: Metabolomic
heterogeneity of pulmonary arterial hypertension. PLoS One.
9:e887272014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiaoying C, Huadan Y, Qingxiao H, Annan Z,
Linlin T and Shiwei D: The effects of DNA methylation on the
homeostasis in vascular diseases. Yi Chuan. 37:221–232. 2015.(In
Chinese). PubMed/NCBI
|
|
10
|
Soubrier F, Chung WK, Machado R, Grünig E,
Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH and
Humbert M: Genetics and genomics of pulmonary arterial
hypertension. J Am Coll Cardiol. 62 25 Suppl:D13–D21. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim JD, Lee A, Choi J, Park Y, Kang H,
Chang W, Lee MS and Kim J: Epigenetic modulation as a therapeutic
approach for pulmonary arterial hypertension. Exp Mol Med.
47:e1752015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pousada G, Baloira A and Valverde D:
Methylation analysis of the BMPR2 gene promoter region in patients
with pulmonary arterial hypertension. Arch Bronconeumol.
52:293–298. 2016.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saco TV, Parthasarathy PT, Cho Y, Lockey
RF and Kolliputi N: Role of epigenetics in pulmonary hypertension.
Am J Physiol Cell Physiol. 306:C1101–C1105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu R, Leslie KL and Martin KA: Epigenetic
regulation of smooth muscle cell plasticity. Biochim Biophys Acta.
1849:448–453. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ospelt C: Epigenetic biomarkers in
rheumatology-the future? Swiss Med Wkly. 146:w143122016.PubMed/NCBI
|
|
16
|
Plant D, Webster A, Nair N, Oliver J,
Smith SL, Eyre S, Hyrich KL, Wilson AG, Morgan AW, Isaacs JD, et
al: Differential methylation as a biomarker of response to
etanercept in patients with rheumatoid arthritis. Arthritis
Rheumatol. 68:1353–1360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zufferey F, Williams FM and Spector TD:
Epigenetics and methylation in the rheumatic diseases. Semin
Arthritis Rheum. 43:692–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu L, Zheng D, Wang L, Jiang D, Liu H, Xu
L, Liao Q, Zhang L, Liu P, Shi X, et al: GCK gene-body
hypomethylation is associated with the risk of coronary heart
disease. Biomed Res Int. 2014:1517232014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bibikova M, Le J, Barnes B,
Saedinia-Melnyk S, Zhou L, Shen R and Gunderson KL: Genome-wide DNA
methylation profiling using Infinium® assay. Epigenomics.
1:177–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bibikova M, Barnes B, Tsan C, Ho V,
Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et
al: High density DNA methylation array with single CpG site
resolution. Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du P, Zhang X, Huang CC, Jafari N, Kibbe
WA, Hou L and Lin SM: Comparison of Beta-value and M-value methods
for quantifying methylation levels by microarray analysis. BMC
Bioinformatics. 11:5872010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bright PD, Mayosi BM and Martin WJ: An
immunological perspective on rheumatic heart disease pathogenesis:
More questions than answers. Heart. 102:1527–1532. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jeffries MA, Dozmorov M, Tang Y, Merrill
JT, Wren JD and Sawalha AH: Genome-wide DNA methylation patterns in
CD4+ T cells from patients with systemic lupus erythematosus.
Epigenetics. 6:593–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernández-Tajes J, Soto-Hermida A,
Vázquez-Mosquera ME, Cortés-Pereira E, Mosquera A, Fernández-Moreno
M, Oreiro N, Fernández-López C, Fernández JL, Rego-Pérez I and
Blanco FJ: Genome-wide DNA methylation analysis of articular
chondrocytes reveals a cluster of osteoarthritic patients. Ann
Rheum Dis. 73:668–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de la Rica L, Urquiza JM, Gómez-Cabrero D,
Islam AB, López-Bigas N, Tegnér J, Toes RE and Ballestar E:
Identification of novel markers in rheumatoid arthritis through
integrated analysis of DNA methylation and microRNA expression. J
Autoimmun. 41:6–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Menzies V, Lyon DE, Archer KJ, Zhou Q,
Brumelle J, Jones KH, Gao G, York TP and Jackson-Cook C: Epigenetic
alterations and an increased frequency of micronuclei in women with
fibromyalgia. Nurs Res Pract. 2013:7957842013.PubMed/NCBI
|
|
27
|
Garcia JJ and Ortega E: Soluble
fractalkine in the plasma of fibromyalgia patients. An Acad Bras
Cienc. 86:1915–1917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Janssens K, Van den Haute C, Baekelandt V,
Lucas S, van Horssen J, Somers V, Van Wijmeersch B, Stinissen P,
Hendriks JJ, Slaets H and Hellings N: Leukemia inhibitory factor
tips the immune balance towards regulatory T cells in multiple
sclerosis. Brain Behav Immun. 45:180–188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thevenon J, Laurent G, Ader F, Laforêt P,
Klug D, Pentiah A Duva, Gouya L, Maurage CA, Kacet S, Eicher JC, et
al: High prevalence of arrhythmic and myocardial complications in
patients with cardiac glycogenosis due to PRKAG2 mutations.
Europace. pii:euw0672016.(Epub ahead of print). View Article : Google Scholar
|
|
30
|
Hedberg-Oldfors C and Oldfors A:
Polyglucosan storage myopathies. Mol Aspects Med. 46:85–100. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kidane YH, Lawrence C and Murali TM:
Computational approaches for discovery of common immunomodulators
in fungal infections: Towards broad-spectrum immunotherapeutic
interventions. BMC Microbiol. 13:2242013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stahl EA, Raychaudhuri S, Remmers EF, Xie
G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A,
et al: Genome-wide association study meta-analysis identifies seven
new rheumatoid arthritis risk loci. Nat Genet. 42:508–514. 2010.
View Article : Google Scholar : PubMed/NCBI
|