Introduction
Myocardial ischemia induced by acute coronary
syndromes remains one of the leading causes of mortality worldwide,
as the heart, which has a high energy demand for its normal
function, is susceptible to ischemia-induced oxygen and glucose
deficiency, which may induce heart damage and even heart failure
(1,2). Restoration of blood to the ischemic
heart, termed reperfusion, either by thrombolysis or primary
percutaneous coronary intervention, is a remedial measure, which
facilitates functional rehabilitation of the heart and attenuates
cardiac apoptosis and necrosis. However, experimental evidence has
largely indicated that timely coronary reperfusion, in turn,
paradoxically harms cardiomyocytes. This phenomenon is referred to
as myocardial ischemia/reperfusion (I/R) injury, for which
particularly the excessive production of reactive oxygen species
(ROS) has been held accountable. Thus, current therapeutic
guidelines highlight the supplementation with anti-oxidative
substances during myocardial I/R (1–3).
Flavonoids are a large group of compounds
(n>4,000) that share a common three-ring structure, but have
different substituents. Although flavonoids have been identified to
have non-caloric and non-nutrient characteristics, numerous members
of this compound class exhibit multiple beneficial bioactivities,
including anti-oxidant, anti-inflammatory, anti-apoptotic and
anticancer properties, and flavonoids are therefore commonly deemed
as promising agents for inhibiting ROS-mediated myocardial damage.
Over the past few decades, this concept has been strongly supported
by numerous studies. Quercetin (Qu), a flavonoid occurring in
various vegetables and traditional Chinese herbal medicines, has
been shown to attenuate myocardial injury induced by myocardial
ischemia-reperfusion (I/R), doxorubicin, and xanthine/xanthine
oxidase (X/XO) through neutralizing ROS and regulating an array of
signaling molecules (4–8). As a widely distributed flavonoid in
fruits, hesperidin has been shown to significantly improve the
inotropic and lusitropic function of the heart, as well as to
reduce left ventricular end-diastolic pressure, the level of
thiobarbituric acid reactive substances as a marker of lipid
peroxidation and the activity of lactate dehydrogenase as a cardiac
injury marker, in an animal model of heart I/R (9). In addition to these flavonoids
available from plants, substantial evidence has indicated that
β-naphthoflavone (β-NF), an artificially synthesized flavonoid,
exerts anti-oxidative action via stimulating the activities of
diverse anti-oxidant enzymes and averting doxorubicin-induced
myocardial damage (10–14). However, certain recent studies have
reported cardiotoxic activities of flavonoids (e.g. Qu and β-NF)
(13,14), although the underlying mechanisms
have remained to be largely elucidated.
Flavonoids have been reported to act as agonists of
the aryl hydrocarbon receptor (Ahr) (15). Ahr is a ligand-activated
transcription factor, which is resident in the cytoplasm in its
latent form, bound to heat shock protein 90 (HSP90). Upon
ligand-mediated activation, Ahr rapidly translocates to the
nucleus, where it dissociates from HSP90 and heterodimerizes with
the Ahr nuclear translocator (ARNT). The Ahr/ARNT complex then
binds to specific recognition sites and initiates the transcription
of target genes of Ahr (16,17). However, the transcriptional
activation regulated by hypoxia-inducible factor (HIF)-1α also
relies on the formation of a complex with ARNT. HIF-1α is an
oxygen-sensitive transcription factor, which exerts a vast array of
physiological functions, enabling cells to adapt to temporary
hypoxia (18,19). Studies have reported that HIF-1α
becomes highly labile under hypoxic conditions induced by
myocardial ischemia and it is allowed to translocate to the nucleus
to trigger transcriptional activation of genes associated with
cardioprotection, such as vascular endothelial growth factor
(VEGF), inducible nitric oxide (NO) synthase (iNOS), erythropoietin
and heme oxygenase-1 (18,19). Based on these findings, the present
study speculated that Ahr activated by certain flavonoids competes
with HIF-1α for binding to ARNT, resulting in the inhibition of
cardioprotection mediated by HIF-1α, which may limit or counteract
the beneficial actions of flavonoids against myocardial I/R.
H9c2 cells initially derived from mitotic rat
embryonic cardiomyocytes have been widely used as a model system to
study cardioprotective and cardiotoxic effects of various
substances in various settings. The present study established a
cellular oxygen glucose deprivation/reoxygenation (OGD/R) model of
myocardial I/R using H9c2 cells and further examined the
cardioprotective roles of Qu and β-NF under these conditions.
Further research efforts were devoted to exploring whether the
interaction between Ahr and HIF-1α mediates the detrimental effects
of Qu and β-NF in myocardial OGD/R.
Materials and methods
Cell culture and treatments
H9c2 cells used in the present study were purchased
from the American Type Culture Collection (Manassas, VA, USA). The
cells were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in a 37°C incubator with 5%
CO2. After reaching 70–80% confluence, the cells were
subjected to oxygen glucose deprivation/reoxygenation (OGD/R) to
simulate I/R injury, as described in the study of Zhao et al
(2), in the absence or presence of
Qu or β-NF. In brief, cells were exposed to hypoxic conditions
(oxygen deprivation, 0.5% O2) for 24 h in culture medium
deprived of glucose and serum, followed by culture under normoxic
conditions (reoxygenation) in normal medium for an additional 24 h.
Qu (0.5, 5 and 50 µM) and β-NF (0.1, 1 and 10 µM; Sigma-Aldrich;
Merck KGaA, Darmstadt Germany) were individually added to the cells
during the entire process of OGD/R.
Immunocytochemical (ICC) assay
An ICC assay was used to analyze the protein levels
of Ahr in the nuclei of H9c2 cells subjected to the abovementioned
treatments. The cells were fixed with 4% paraformaldehyde for 15
min and blocked with PBS containing 0.3% Triton X-100 and 5% bovine
serum albumin (w/v) (Gibco; Thermo Fisher Scientific, Inc.) for 1 h
at room temperature. Subsequently, the cells were incubated with
primary antibody specific for Ahr (1:500 dilution; cat. no. ab2770;
Abcam, Cambridge, UK) for 2 h at room temperature prior to
incubation with the secondary fluorescent-labeled antibody
(A-21202; Alexa Fluor 488; Invitrogen; Thermo Fisher Scientific,
Inc.) at room temperature for 1 h. DAPI (1:1,000 dilution;
Invitrogen; Thermo Fisher Scientific, Inc.) and
fluorescence-quenching agent (TF-B21; Weifang Greatland Chemicals
Co., Ltd, Shandong, China) were added to the cells in the dark for
5 min, followed by analysis with a fluorescence microscope (Nikon,
Tokyo, Japan) and analysis of the images using DP2-BSW software
(Olympus, Tokyo, Japan).
Knockdown of Ahr in H9c2 cells
Small interfering RNA (siRNA) targeting Ahr
(siRNA-Ahr) was synthesized by GenePharma Co., Ltd. (Shanghai,
China). siRNA-Ahr was transfected into H9c2 cells using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. H9c2 cells with Ahr
knockdown were further subjected to OGD/R in the absence or
presence of Qu or β-NF.
Determination of apoptotic rate
The apoptotic rate of H9c2 cells was assessed by
using an Annexin V-fluorescein isothiocyanate/propidium iodide kit
(Kaiji Biological Inc., Nanjing, China) according to the
manufacturer's protocol. A dual laser flow cytometer (Becton
Dickinson, San Jose, CA, USA) with ModFit LT software (Verity
Software House, Topsham, ME, USA) was used to determine the
apoptotic rate.
Lactate hydrogenase (LDH) leakage
assay
The amount of LDH in the culture medium was examined
with using a LDH Activity Assay kit (Beyotime Institute of
Biotechnology, Haimen, China). After the respective treatments,
cell culture medium was collected and transferred to a 96-well
plate. LDH reaction mix was added to each well, and the plates were
incubated for 30 min at room temperature. Finally, the optical
density was determined at 450 nm using an ELISA plate reader (Model
550; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Measurement of intracellular ROS and
cell total anti-oxidant capacity (TAOC)
Intracellular ROS levels in H9c2 cells were
quantified using a Reactive Oxygen Species Assay kit (Beyotime
Institute of Biotechnology). After washing with PBS, the cells were
suspended in 2′,7′-dichlorofluorescin diacetate (DCFH-DA) solution
(10 µM) at 107/ml and incubated at 37°C for 20 min. The
fluorescence intensity of DCFH-DA in the cells was detected by a
fluorospectrophotometer (F-4000; Hitachi, Ltd., Tokyo, Japan).
The azino-diethyl-benzthiazoline sulfate (ABTS)
method was adopted to examine the TAOC of H9c2 cells. Incubation
with ABTS with H2O2 and a peroxidase
(metmyoglobin) (provided by ABTS detection kit from Beyotime
Institute of Biotechnology) results in the production of a
blue-green radical cation ABTS+. Anti-oxidants contained
in the H9c2 cells suppress this color production proportionally to
the cells' TAOC. The system was standardized using Trolox (provided
by ABTS detection kit), a water-soluble vitamin E analogue. The
results were expressed as µmol Trolox equivalent/protein
concentration of the H9c2 cells.
Intracellular NO measurement
NO in the H9c2 cells was detected using the NO
Detection kit (Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. In brief, the level of the NO
derivative nitrite was determined via the Griess reaction. A
standard curve was generated using NaNO2 mixed with
Griess reagent. After 15 min, optical density was read using a
microplate reader at 540 nm.
Western blot analysis
Total proteins were extracted from H9c2 cells with a
cell lysis reagent (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's instructions and protein was quantified using the
bicinchoninic acid method (Beyotime Institute of Biotechnology).
The extracted proteins (20 µg per lane) were separated by 10–15%
SDS-PAGE and transferred onto nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked in 5%
non-fat milk in Tris-buffered saline/0.1% Tween-20 for 2 h prior to
immunoblotting at 4°C overnight with the following antibodies:
Anti-Ahr, anti-HIF-1α (1:300 dilution, cat. no. ab463; Abcam),
anti-Caspase-3 (1:500 dilution, cat. no. ab90437; Abcam), iNOS
(1:500 dilution, cat. no. ab15323; Abcam), VEGF (1:800 dilution,
cat. no. bs-1665R; Bioss, Beijing, China) and GAPDH (1:1,000
dilution, cat. no. sc-365062; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA). Membranes were then incubated with horseradish
peroxidase-conjugated secondary antibody (1:20,000 dilution; cat.
no. A9309, Sigma-Aldrich, Merck KGaA; and cat. no. ab97051, Abcam)
for 2 h at room temperature. The intensities of the bands were
quantified using an enhanced chemiluminesence detection kit
(Pierce; Thermo Fisher Scientific, Inc.) and an image analysis
system (ProteinSimple; Bio-Techne, Minneapolis, MN, USA).
Co-immunoprecipitation (Co-IP)
assay
Nucleoprotein in H9c2 cells was extracted with a
CelLytic™ NuCLEAR™ Extraction kit
(Sigma-Aldrich; Merck KGaA) and incubated ARNT primary antibody
(1:500 dilution, cat. no. sc-5580; Santa Cruz Biotechnology, Inc.)
at 4°C for 60 min with gentle mixing. Subsequently, 20 µl Protein
A/G Plus-Agarose beads (Thermo Fisher Scientific, Inc.) was added,
followed by incubation at 4°C overnight. The mixture was
centrifuged at 500 × g for 5 min at 4°C. The supernatant was
discarded and the Co-IP products were washed three times with PBS.
After the final wash, the precipitates were re-suspended in 40 µl
sample buffer and detected by western blotting with anti-Ahr (1:200
dilution) and anti-HIF-1α antibodies (1:200 dilution).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical analysis was performed using SPSS v12.0
software (SPSS, Inc., Chicago, IL, USA). One-way analysis of
variance with Scheffe's post-hoc testing was used for multiple
comparisons between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Qu and β-NF drive the translocation of
Ahr from cytoplasm to cell nucleus
Ahr is a receptor mainly present in the cytoplasm in
its latent form, while it translocates from the cytoplasm into the
cell nucleus to trigger target gene expression once activated by
its ligands. An immunocytochemical assay demonstrated that H9c2
cells subjected OGD/R alone showed no significant change in Ahr
fluorescence intensity (FI) in the cell nucleus (Fig. 1). Supplementation with Qu at 5 or 50
µM during the OGD/R process increased Ahr FI in the cell nucleus
(P<0.05 vs. control group). Furthermore, Ahr FI in the cell
nucleus was dose-dependently increased by β-NF within the tested
concentrations in the OGD/R process, with 10 µM β-NF causing the
greatest increase (P<0.05 vs. control group). Based on these
findings, 5 µM Qu and 10 µM β-NF were selected as the treatment
concentrations used in subsequent assays.
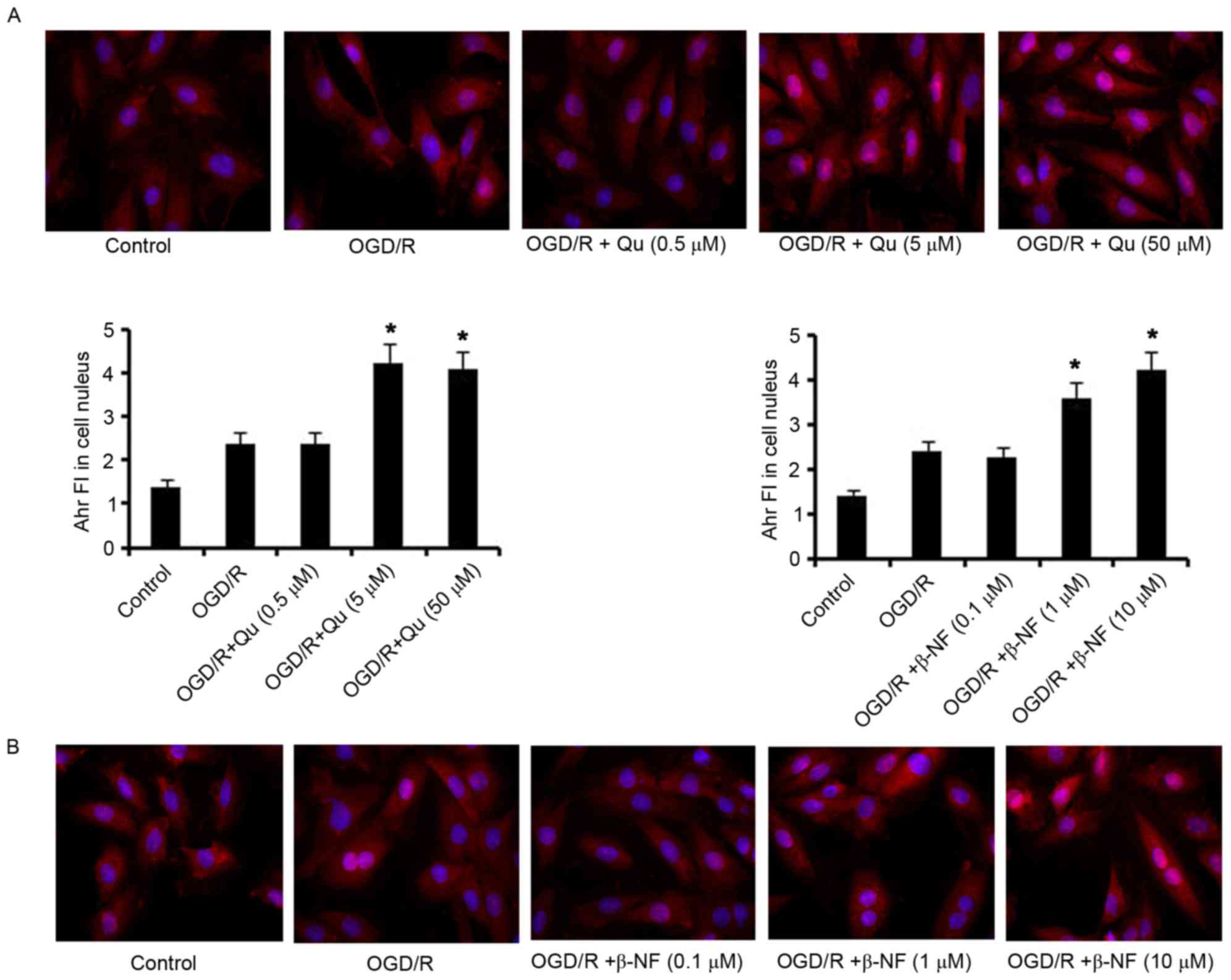 | Figure 1.Qu and β-NF lead to the translocation
of Ahr from cytoplasm to cell nucleus. H9c2 cells were exposed to
hypoxic conditions (oxygen deprivation, 0.5% O2) for 24
h in culture medium deprived of glucose and serum, followed by
culture under normoxic conditions (reoxygenation) in normal medium
for an additional 24 h. (A) Qu (0.5, 5 and 50 µM) and (B) β-NF
(0.1, 1 and 10 µM) were added individually to the cells during the
whole process of OGD/R. An immunocytochemical assay was used to
analyze the protein level of Ahr in the nuclei in treated H9c2
cells. Magnification, ×400. *P<0.05 vs. control. Ahr, aryl
hydrocarbon receptor; Qu, quercetin; β-NF, β-naphthoflavone; OGD/R,
oxygen glucose deprivation/reoxygenation; FI, fluorescence
intensity. |
Qu and β-NF inhibit cell death, LDH
leakage and caspase-3 activation caused by OGD/R
Exposure of H9c2 cells to OGD/R induced an increase
in the apoptotic rate (P<0.05 vs. control group, Fig. 2A), which was individually decreased
by 5 µM Qu and 10 µM β-NF (P<0.05 vs. OGD/R group). Ahr
knockdown in H9c2 cells attenuated the protective effect of 10 µM
β-NF against cell death (P<0.05), but barely influenced the
protective effect of 5 µM Qu. LDH leakage is an important indicator
of myocardial insult induced by I/R. Treatment with OGD/R augmented
LDH leakage from H9c2 cells (P<0.01 vs. control group; Fig. 2B), whereas this action was
individually inhibited by 5 µM Qu and 10 µM β-NF (P<0.05 vs.
OGD/R group). The protective effect of 5 µM Qu and 10 µM β-NF
against LDH leakage was diminished after Ahr knockdown (P<0.05).
As a critical apoptotic marker, caspase-3 activity is positively
correlated with the cleaved caspase-3 protein level. An increase in
the levels of cleaved caspase-3 protein in H9c2 cells was observed
after OGD/R compared with that in the control group (P<0.05,
Fig. 2C). Supplementation with 5 µM
Qu during the OGD/R process inhibited the increase of cleaved
caspase-3 in H9c2 cells (P<0.05 vs. OGD/R group). Ahr knockdown
did not influence the inhibitory effect. Addition of 10 µM β-NF to
H9c2 the cells diminished the upregulation of cleaved caspase-3
induced by OGD/R (P<0.05 vs. OGD/R group). However, Ahr
knockdown attenuated the inhibition of cleaved caspase-3 by β-NF
(P<0.05 vs. β-NF group without knockdown).
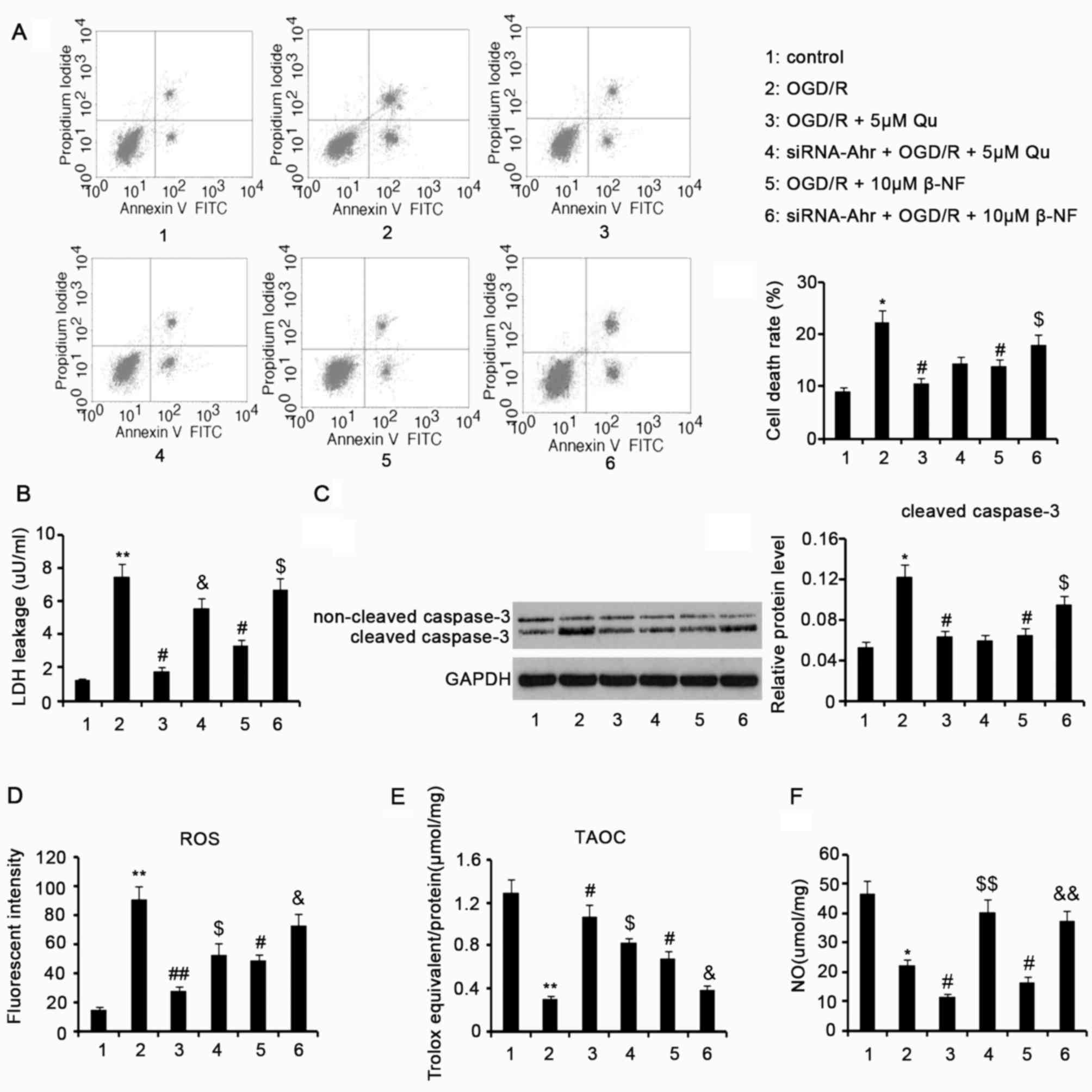 | Figure 2.Qu and β-NF protect cells from lethal
injury and decrease ROS in cells. H9c2 cells with or without Ahr
knockdown were exposed to hypoxic conditions (oxygen deprivation,
0.5% O2) for 24 h in culture medium deprived of glucose
and serum, followed by culture under normoxic conditions
(reoxygenation) in normal medium for an additional 24 h. 5 µM Qu or
10 µM β-NF was added individually to the cells during the entire
process of OGD/R. (A) The cell death rate was assessed by flow
cytometry. The total number of cells in each group was
2×107. (B) Lactate hydrogenase leakage was assessed by a
colorimetric assay. (C) Cleaved caspase-3 protein levels were
assessed by western blot analysis. (D) Intracellular ROS, (E) TAOC
of the cells and (F) NO levels were assessed after the treatments.
Groups: 1, Control; 2, OGD/R; 3, OGD/R + 5 µM Qu; 4, siRNA-Ahr +
OGD/R + 5 µM Qu; 5, OGD/R + 10 µM β-NF; 6, siRNA-Ahr + OGD/R + 10
µM β-NF. *P<0.05, **P<0.01 vs. control group;
#P<0.05, ##P<0.01 vs. OGD/R group;
$P<0.05 and $$P<0.01 vs. Qu group
without Ahr knockdown; &P<0.05 and
&&P<0.01 vs. β-NF group without Ahr
knockdown. ROS, reactive oxygen species; Qu, quercetin; β-NF,
β-naphthoflavone; OGD/R, oxygen glucose deprivation/reoxygenation;
Ahr, aryl hydrocarbon receptor; siRNA-Ahr, small interfering RNA
against Ahr; FITC, fluorescein isothiocyanate; TAOC, total
antioxidant capacity; NO, nitric oxide. |
Qu and β-NF inhibit intracellular ROS
production and decreases in cell TAOC during OGD/R
H9c2 cells subjected to OGD/R showed increased
intracellular ROS levels compared with those in the control
(P<0.01; Fig. 2D).
Supplementation with 5 µM Qu during the OGD/R process lowered the
ROS in H9c2 cells (P<0.01 vs. OGD/R group), but Ahr knockdown
partly attenuated the anti-oxidative function (P<0.05 vs. Qu
group without knockdown). Decreased ROS were also observed in H9c2
cells when 10 µM β-NF was added during the OGD/R process (P<0.05
vs. OGD/R group). However, Ahr knockdown reversed the
anti-oxidative function (P<0.05 vs. β-NF group without
knockdown). OGD/R decreased the cell TAOC relative to that in the
control (P<0.01), which was attenuated by 5 µM Qu and 10 µM β-NF
(P<0.05 vs. OGD/R group; Fig.
2E). Ahr knockdown in H9c2 cells inhibited the effect of Qu and
β-NF on increasing TAOC (both P<0.05).
NO content in H9c2 cells after various
treatments
The NO content in H9c2 cells was decreased after
H9c2 cells were subjected to OGD/R compared with that in the
control cells (P<0.05; Fig. 2F).
Supplementation with 5 µM Qu or 10 µM β-NF in the OGD/R process
resulted in a further decrease in NO content compared with that in
the OGD/R-treated group (P<0.05). Ahr knockdown conversely
increased NO content in H9c2 cells that were subjected to OGD/R in
the presence of Qu and β-NF, compared with the cells without Ahr
knockdown (both P<0.01).
Target protein levels after various
treatments
Western blot assay revealed that HIF-1α protein
levels were upregulated after H9c2 cells were subjected to OGD/R
(P<0.05 vs. control; Fig. 3A). In
comparison to the OGD/R-treated group, supplementation with 5 µM Qu
or 10 µM β-NF in the process of OGD/R did not significantly affect
HIF-1α protein levels regardless of whether Ahr knockdown was
performed. iNOS protein levels in H9c2 cells were significantly
decreased by treatment with OGD/R (P<0.05). Addition of 5 µM Qu
or 10 µM β-NF to cells in the process of OGD/R further decreased
iNOS protein levels (P<0.05 vs. OGD/R group), which was reversed
by Ahr knockdown (P<0.01 vs. the Qu and β-NF groups without
knockdown). OGD/R treatment promoted VEGF protein expression in
H9c2 cells (P<0.05 vs. control group), which was significantly
inhibited by supplementation with 5 µM Qu or 10 µM β-NF (P<0.05
vs. OGD/R group). However, Ahr knockdown impaired the inhibited
effect of Qu and β-NF on VEGF expression (P<0.05 and P<0.01,
respectively).
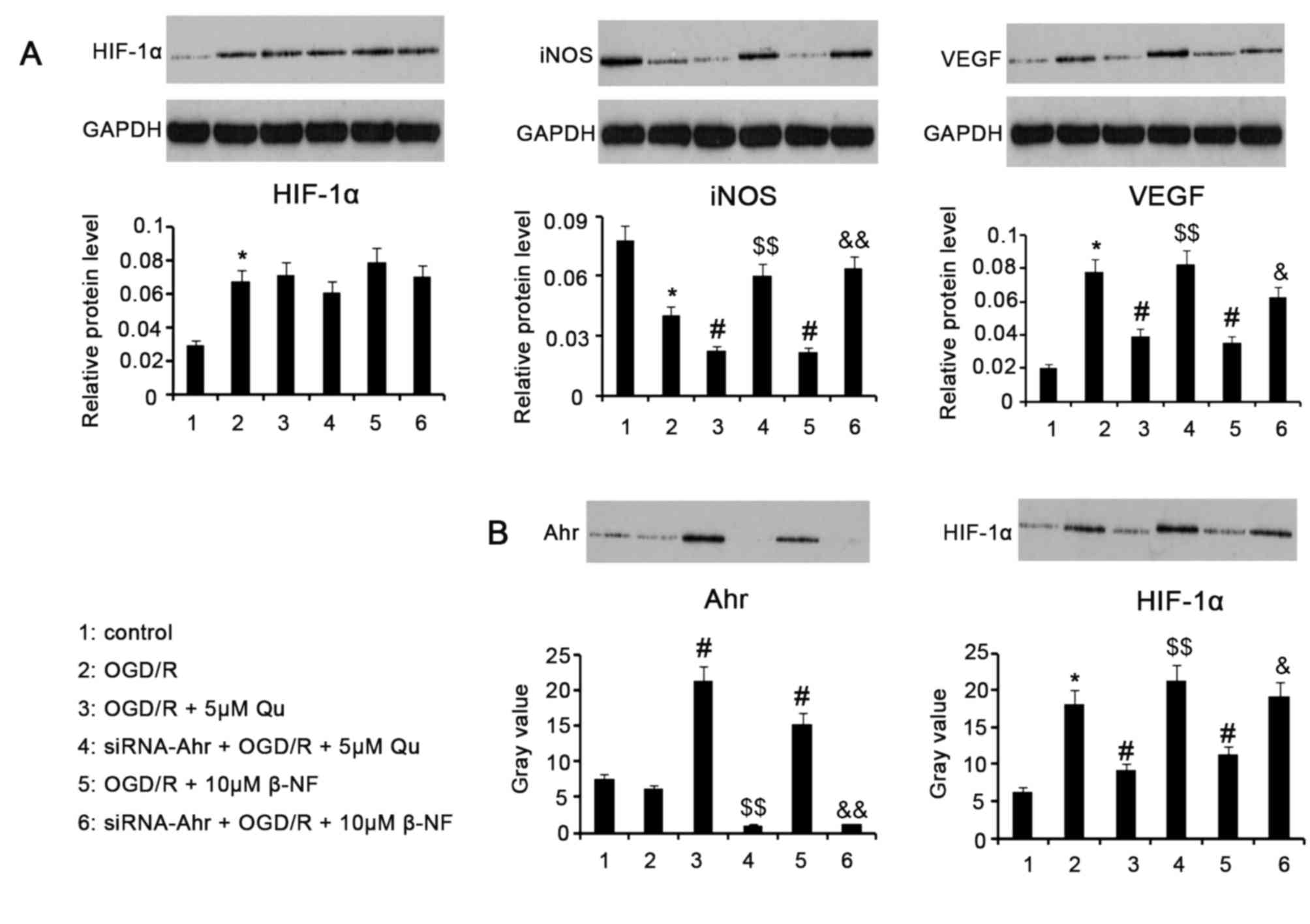 | Figure 3.Target protein levels after various
treatments. H9c2 cells with or without Ahr knockdown were exposed
to hypoxic conditions (oxygen deprivation, 0.5% O2) for
24 h in culture medium deprived of glucose and serum, followed by
culture under normoxic conditions (reoxygenation) in normal medium
for an additional 24 h. 5 µM Qu and 10 µM β-NF were added
individually to the cells during the whole process of OGD/R. (A)
Western blot analysis was used for assessing the levels of HIF-1α,
iNOS and VEGF in the treated cells. (B) A co-immunoprecipitation
assay was used to test the interaction between Ahr and HIF-1α in
terms of ARNT binding in the nuclei of treated cells. Nucleoprotein
extracted from H9c2 cells was incubated with ARNT primary antibody
and then subjected to western blotting with anti-Ahr or anti-HIF-1α
antibodies. Groups: 1, Control; 2, OGD/R; 3, OGD/R + 5 µM Qu; 4,
siRNA-Ahr + OGD/R + 5 µM Qu; 5, OGD/R + 10 µM β-NF; 6, siRNA-Ahr +
OGD/R + 10 µM β-NF. *P<0.05 vs. control group;
#P<0.05 vs. OGD/R group; $P<0.05,
$$P<0.01 vs. Qu group without Ahr knockdown;
&P<0.05, &&P<0.01 vs. β-NF
group without Ahr knockdown. Qu, quercetin; β-NF, β-naphthoflavone;
OGD/R, oxygen glucose deprivation/reoxygenation; Ahr, aryl
hydrocarbon receptor; siRNA-Ahr, small interfering RNA against Ahr;
ARNT, Ahr nuclear translocator; iNOS, inducible nitric oxide
synthase; HIF, hypoxia-inducible factor; VEGF, vascular endothelial
growth factor. |
Ahr competes with HIF-1α for combining
with ARNT after stimulation with Qu and β-NF
The Co-IP assay indicated that OGD/R promoted the
binding of ARNT to HIF-1α (P<0.05 vs. control group, Fig. 3B), but had little effect on the
binding of ARNT to Ahr. Supplementation with Qu and β-NF during
OGD/R promoted the binding of ARNT to Ahr (both P<0.05 vs. OGD/R
group), but decreased the binding of ARNT to HIF-1α (both P<0.05
vs. OGD/R group). However, knockdown of Ahr in H9c2 led to a
decrease in the binding of ARNT to Ahr (P<0.01 vs. Qu and β-NF
groups without Ahr knockdown) and an increase in the binding of
ARNT to HIF-1α (P<0.01 vs. Qu group without Ahr knockdown and
P<0.05 vs. β-NF group without Ahr knockdown) in the presence of
Qu and β-NF. These data in combination with results from the ICC
assay indicate that Qu and β-NF promote nuclear translocation of
Ahr; Ahr binds to ARNT in the nucleus, resulting in reduced binding
of ARNT to HIF-1α.
Discussion
Previous studies have extensively reported that ROS
are largely produced in myocardial ischemia as well as reperfusion,
although the underlying mechanisms are probably different (3). Continuous deficiency of oxygen and
glucose during myocardial ischemia disrupts mitochondrial
homeostasis and metabolism, facilitating the conversion of
O2 to O2− and other ROS due to
increased electron leakage. Timely reperfusion indeed eases
ischemic injury and salvages viable myocardium, while NADPH
oxidases, lipoxygenase and xanthine oxidase are activated in
response to the reperfusion, which are responsible for the
generation of most of the ROS in this process. The present study
established a cellular OGD/R model of I/R and showed that ROS was
markedly and consistently elevated in these treated H9c2 cells
(3). The significantly increased ROS
leads to a huge consumption of anti-oxidative substances and
suppresses the activities of certain anti-oxidant enzymes,
resulting in an attenuated cell TOAC. In addition to ATP depletion
and Ca2+ overload, the disruption of the oxidative and
anti-oxidative balance has in OGD/R been associated with myocardial
damage and death, (3). In agreement
with previous studies, the results of the present study showed that
the apoptotic rate, LDH leakage and caspase-3 activity were
increased in parallel with the significantly elevated ROS.
The anti-oxidative properties of Qu and β-NF deserve
to be acknowledged, based on the results of the present and
previous studies. In the present study, supplementation with Qu or
β-NF during the OGD/R process notably diminished ROS in H9c2 cells
and reinforced the cell TOAC. Several studies have demonstrated
that Qu has the capability to scavenge superoxide anions, singlet
oxygen and lipid peroxy radicals in vitro and in animal
models (4–8). β-NF has been shown to strengthen the
activities of anti-oxidative enzymes, such as glutathione
peroxidase, quinone oxidoreductase 1, glutathione transferase and
heme oxygenase 1, and to repress NADPH oxidases that are
ROS-producing enzymes, thereby having an important anti-oxidant
role (10–14). Qu and β-NF caused decreases in ROS in
H9c2 cells, thus protecting the cells from death and impairment
resulting from exposure to OGD/R. An accidental discovery of the
present study was that Ahr knockdown notably attenuated the
anti-oxidative action of β-NF, suggesting that Ahr mediated the
anti-oxidative action. Slightly different from β-NF, the
anti-oxidative capacity of Qu was partly decreased by Ahr
knockdown, which suggested that anti-oxidative function of Qu is at
least partly dependent on Ahr signaling.
The cardioprotective actions mediated by HIF-1α in
myocardial I/R have been partly elucidated. The stability of HIF-1α
is regulated by the HIF-prolyl hydroxylases domain (PHD) that
targets it for polyubiquitination and proteosomal degradation. The
hypoxia induced by myocardial ischemia inhibits the activity of
HIF-PHD, thereby allowing HIF-1α to accumulate and translocate to
the nucleus, where it binds to ARNT and regulates the transcription
of certain hypoxia-responsive genes (20–22).
Certain experimental studies have shown that genetic or
pharmacological stabilization of HIF-1α protects the heart against
the detrimental effects of acute I/R injury by enhancing iNOS, VEGF
and B-cell lymphoma-2 expression and restricting nuclear
factor-κB-dependent gene expression (20–22). In
the present study, HIF-1α protein levels in H9c2 cells were
markedly elevated in response to OGD/R, which represents a
self-protective mechanism of myocardial H9c2 cells in response to
this challenge. Although no significant difference in HIF-1α
protein levels was observed with Qu and β-NF addition during OGD/R,
the ICC and Co-IP assays revealed that Qu and β-NF promoted the
translocation of Ahr from the cytoplasm into the cell nucleus,
where Ahr dimerized with ARNT to significantly decrease the amount
of ARNT that binds to HIF-1α. Ahr is a cytosolic ligand-activated
transcription factor that can be activated by a class of
flavonoids. Activated Ahr translocates into the cell nucleus and
binds to ARNT to form the Ahr/ARNT complex, and then binds to
specific recognition sites in its target genes. As a likely
consequence of ARNT binding to Ahr present in large amounts but
rarely to HIF-1α, the cardioprotection mediated by HIF-1α is
attenuated.
In accordance with this presumption, iNOS and VEGF
protein levels together with the NO content were significantly
decreased in H9c2 cells after treatment with Qu or β-NF. Knockdown
of Ahr in H9c2 cells in the presence of Qu or β-NF increased
binding of ARNT to HIF-1α, which was accompanied with marked
increases in iNOS and VEGF protein levels as well as NO formation.
It has been well-documented that NO exerts robust cardioprotective
effects against I/R injury. NO is a gaseous signaling molecule that
participates in a wide variety of cardiovascular functions,
including vasodilatation, neovascularization, scavenging of ROS and
regulation of the cardiac immune response. Studies have revealed a
reduction in the bioavailability of NO in the reperfusion phase,
while interventions that can increase NO formation have a
significant therapeutic effect against myocardial I/R injury
(23–25). The importance of VEGF in the
inhibition of the progression of myocardial I/R injury has been
recognized for several years. Several substances showing protection
against myocardial I/R injury have been demonstrated to act in a
VEGF-dependent manner (26). VEGF
gene therapy for the purpose of promoting therapeutic angiogenesis
and recovering the pressure developed in the left ventricle has now
been advanced as an alternative treatment for myocardial I/R
(22,26). It is therefore likely that
flavonoid-induced decreases in NO and VEGF formation probably
exacerbate myocardial I/R injury.
In conclusion, the present study confirmed the
anti-oxidative and protective effects of Qu and β-NF in the OGD/R
process of H9c2 cells. More importantly, the present study revealed
that Qu and β-NF attenuated the HIF-1α-mediated cardioprotection
through activating Ahr. The results provided by the present study
suggest a dual character of certain flavonoids in the process of
myocardial I/R.
References
|
1
|
Wu WY, Wang WY, Ma YL, Yan H, Wang XB, Qin
YL, Su M, Chen T and Wang YP: Sodium tanshinone IIA silate inhibits
oxygen-glucose deprivation/recovery-induced cardiomyocyte apoptosis
via suppression of the NF-κB/TNF-α pathway. Br J Pharmacol.
169:1058–1071. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao D, Li Q, Huang Q, Li X, Yin M, Wang Z
and Hong J: Cardioprotective effect of propofol against oxygen
glucose deprivation and reperfusion injury in H9c2 cells. Oxid Med
Cell Longev. 2015:1849382015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
ischemia-reperfusion injury. Biomed Res Int. 2015:8649462015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen JY, Hu RY and Chou HC:
Quercetin-induced cardioprotection against doxorubicin
cytotoxicity. J Biomed Sci. 20:952013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Y, Zhang ZZ, Wu Y, Ke JJ, He XH and
Wang YL: Quercetin postconditioning attenuates myocardial
ischemia/reperfusion injury in rats through the PI3K/Akt pathway.
Braz J Med Biol Res. 46:861–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dong Q, Chen L, Lu Q, Sharma S, Li L,
Morimoto S and Wang G: Quercetin attenuates doxorubicin
cardiotoxicity by modulating Bmi-1 expression. Br J Pharmacol.
171:4440–4454. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ozbek N, Bali EB and Karasu C: Quercetin
and hydroxytyrosol attenuates xanthine/xanthine oxidase-induced
toxicity in H9c2 cardiomyocytes by regulation of oxidative stress
and stress-sensitive signaling pathways. Gen Physiol Biophys.
34:407–414. 2015.PubMed/NCBI
|
|
8
|
Agrawal YO, Sharma PK, Shrivastava B, Ojha
S, Upadhya HM, Arya DS and Goyal SN: Hesperidin produces
cardioprotective activity via PPAR-γ pathway in ischemic heart
disease model in diabetic rats. PLoS One. 9:e1112122014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nannelli A, Rossignolo F, Tolando R,
Rossato P, Longo V and Gervasi PG: Effect of beta-naphthoflavone on
AhR-regulated genes (CYP1A1, 1A2, 1B1, 2S1, Nrf2, and GST) and
antioxidant enzymes in various brain regions of pig. Toxicology.
265:69–79. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morrissy S, Strom J, Purdom-Dickinson S
and Chen QM: NAD (P)H:quinone oxidoreductase 1 is induced by
progesterone in cardiomyocytes. Cardiovasc Toxicol. 12:108–114.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Higgins LG and Hayes JD: Mechanisms of
induction of cytosolic and microsomal glutathione transferase (GST)
genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev.
43:92–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reed JR, Cawley GF and Backes WL:
Inhibition of cytochrome P450 1A2-mediated metabolism and
production of reactive oxygen species by heme oxygenase-1 in rat
liver microsomes. Drug Metab Lett. 5:6–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Daubney J, Bonner PL, Hargreaves AJ and
Dickenson JM: Cardioprotective and cardiotoxic effects of quercetin
and two of its in vivo metabolites on differentiated h9c2
cardiomyocytes. Basic Clin Pharmacol Toxicol. 116:96–109. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zordoky BN and El-Kadi AO:
2,3,7,8-Tetrachlorodibenzo-p-dioxin and beta-naphthoflavone induce
cellular hypertrophy in H9c2 cells by an aryl hydrocarbon
receptor-dependant mechanism. Toxicol In Vitro. 24:863–871. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vrba J, Kren V, Vacek J, Papouskova B and
Ulrichova J: Quercetin, quercetin glycosides and taxifolin differ
in their ability to induce AhR activation and CYP1A1 expression in
HepG2 cells. Phytother Res. 26:1746–1752. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smith KJ, Murray IA, Tanos R, Tellew J,
Boitano AE, Bisson WH, Kolluri SK, Cooke MP and Perdew GH:
Identification of a high-affinity ligand that exhibits complete
aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther.
338:318–327. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Narayanan GA, Murray IA, Krishnegowda G,
Amin S and Perdew GH: Selective aryl hydrocarbon receptor
modulator-mediated repression of CD55 expression induced by
cytokine exposure. J Pharmacol Exp Ther. 342:345–355. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsai CH, Li CH, Liao PL, Cheng YW, Lin CH,
Huang SH and Kang JJ: NcoA2-dependent inhibition of HIF-1α
activation is regulated via AhR. Toxicol Sci. 48:17–30. 2015.
|
|
19
|
Ong SG and Hausenloy DJ: Hypoxia-inducible
factor as a therapeutic target for cardioprotection. Pharmacol
Ther. 136:69–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adluri RS, Thirunavukkarasu M, Dunna NR,
Zhan L, Oriowo B, Takeda K, Sanchez JA, Otani H, Maulik G, Fong GH
and Maulik N: Disruption of hypoxia-inducible transcription
factor-prolyl hydroxylase domain-1 (PHD-1-/-) attenuates ex vivo
myocardial ischemia/reperfusion injury through hypoxia-inducible
factor-1α transcription factor and its target genes in mice.
Antioxid Redox Signal. 15:1789–1797. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bandarra D, Biddlestone J, Mudie S, Müller
HA and Rocha S: HIF-1α restricts NF-κB-dependent gene expression to
control innate immunity signals. Dis Model Mech. 8:169–181. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Poynter JA, Manukyan MC, Wang Y, Brewster
BD, Herrmann JL, Weil BR, Abarbanell AM and Meldrum DR: Systemic
pretreatment with dimethyloxalylglycine increases myocardial HIF-1α
and VEGF production and improves functional recovery after acute
ischemia/reperfusion. Surgery. 150:278–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Siu KL, Lotz C, Ping P and Cai H: Netrin-1
abrogates ischemia/reperfusion-induced cardiac mitochondrial
dysfunction via nitric oxide-dependent attenuation of NOX4
activation and recoupling of NOS. J Mol Cell Cardiol. 78:174–185.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alánová P, Kolář F, Ošťádal B and Neckář
J: Role of NO/cGMP signaling pathway in cardiac ischemic tolerance
of chronically hypoxic rats. Physiol Res. 64:783–787.
2015.PubMed/NCBI
|
|
25
|
Totzeck M, Hendgen-Cotta U and Rassaf T:
Concepts of hypoxic NO signaling in remote ischemic
preconditioning. World J Cardiol. 7:645–651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bir SC, Pattillo CB, Pardue S, Kolluru GK,
Shen X, Giordano T and Kevil CG: Nitrite anion therapy protects
against chronic ischemic tissue injury in db/db diabetic mice in a
NO/VEGF-dependent manner. Diabetes. 63:270–281. 2014. View Article : Google Scholar : PubMed/NCBI
|














