Introduction
Osteoporosis is a multi-gene disease. It has been
shown that polymorphisms in the osteoprotegerin (OPG) gene are
closely related to its occurrence. Because of the anti-osteoclast
activity of OPG, it has become a candidate gene for the treatment
of osteoporosis (1–3). At present, many studies have shown that
polymorphisms in the OPG gene promoter and introns primarily
involve the sites 163A-G, 209G-A, 245T-G, 889C-T, 950T-C, 1181G-C,
and 6890A-C. A previous study found that 163, 209, 245 and 1181
site polymorphisms are associated with lower bone mineral density
(BMD) or bone fracture, among which, the correlation of the 245
site with the pathological changes is the largest. In contrast, the
889, 950 and 6890 site polymorphisms are not associated with
osteoporosis (4). Additionally,
Japanese scholars found that the T-C polymorphism at the 223 site
upstream of the OPG transcription start region is related to lower
BMD (5). Another study reported that
the 445 C-T polymorphism is related to Paget bone disease. During
our study, we identified the genetic polymorphism, g.27563G>A,
within the fifth exon of the OPG gene by created restriction
site-polymerase chain reaction (CRS-PCR). The potential
relationship between this genetic polymorphism and both BMD and
osteoporosis was analyzed. The results showed that there was a
significant correlation between BMD, osteoporosis, and the genetic
polymorphism in Chinese menopausal women. The female BMD associated
with the GG genotype was significantly higher than that of the GA
and AA genotypes. Therefore, the A-allele was a risk factor for BMD
and osteoporosis. However, the specific association between OPG
gene polymorphisms and osteoporosis remains unclear.
Materials and methods
Experimental instruments
Electronic micro-balance (Changsha Xiangping Science
and Technology Development Co., Ltd., Changsha, China); ice maker
(Sanyo Electric Co., Ltd., Moriguchi, Japan); −70°C ultra-low
temperature freezer (Forma Scientific, Inc., Marietta, OH, USA);
thermostat-controlled water-bath (Ningbo Xinzhi Biotechnology Co.,
Ltd., Ningbo, China); oscillation mixing device (Shanghai Huayun
Analytical Instrument Co., Ltd., Shanghai, China); cryogenic
supercentrifuge (DuPont, Wilmington, DE, USA); table concentrator
(Barnstead International Co., Ltd., Boston, MA, USA); UV
spectrophotometer (Hitachi, Tokyo, Japan); agarose gel
electrophoresis apparatus (Beijing Liuyi Instrument Factory,
Beijing, China); UV gel imaging system (Bio-Rad, Hercules, CA,
USA); DNA Engine PCR instrument (University of Leicester, London,
UK); clean bench (Beijing Semiconductor Equipment Factory, Beijing,
China); CO2 incubator (Thermo Electron Corp., Waltham,
MA, USA).
Experimental materials
Human embryonic kidney (HEK)293 cells were purchased
from American Type Culture Collection (ATCC, Manassas, VA, USA),
and cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% fetal bovine serum (FBS) to maintain adherent growth
of cells.
RAW264.7 macrophages were from ATCC, and cultured in
DMEM (containing 10% FBS, 2 mmol/l L-glutamine, 100 IU/ml
penicillin, and 100 µg/ml streptomycin), under 5% CO2,
37°C, and saturated humidity. Culture medium was changed every two
days. Adherent cells were digested and subcultured when they
reached 80–90% confluence.
Reagents
Lipofectamine 2000 and α-minimal essential medium
(α-MEM) (both from Invitrogen, Carlsbad, CA, USA); Fast Mutagenesis
system (Beijing TransGen Biotech Co., Ltd., Beijing, China); TRIzol
reagent (Invitrogen); MTT cell proliferation assay kit (Nanjing
Kaiji Biology Company, Nanjing, China); PCR kit (Beijing TransGen
Biotech Co., Ltd.).
Construction of the mutant-type
plasmid
The pcDNA3.0-OPG plasmid (wild-type, GG type) was
synthesized and provided by Kangwei Century Co. (Beijing, China).
The mutant-type plasmid (GA type) was obtained with the fast
site-directed mutagenesis kit (Fast Mutagenesis system). The PCR
site-directed mutagenesis method was provided by Beijing TransGen
Biotech Co., Ltd. The primers were designed according to the primer
design principles of the Fast Mutagenesis system, and all primers
were synthesized by Invitrogen Biological Technology Co., Ltd.
(Shanghai, China).
Cell transfection
The wild-type OPG expression vector and mutant-type
OPG expression vector were transfected into HEK293 cells as
follows: i) HEK293 cells were seeded in 24-well plates, with
2×104 cells per well; ii) Lipofectamine 2000 reagent
diluted with medium, was added to each well and left to stand for
10 min at room temperature; iii) the wild-type OPG expression
vector or mutant-type OPG expression vector were added, followed by
addition of an appropriate amount of medium. Cells were then left
to stand for 10 min at room temperature; and iv) cells were then
left to incubate at 5% CO2 and 37°C for 5 h, after which
the medium was replaced with fresh complete medium. Cells were then
left to incubate for 36–48 h.
RNA extraction
A total of 0.4 ml TRIzol reagent was added to
harvested cells. The samples were mixed and left to stand at room
temperature for 5 min. Chloroform (1/5 of the volume of TRIzol) was
added, mixed evenly, and left to stand for 5 min at room
temperature. Subsequently, the solution was centrifuged at low
temperature for 15 min at 12,000 × g. The supernatants were
collected and transferred to clean centrifuge tubes. Equal volumes
of isopropanol were then added, mixed evenly, and incubated for 10
min at room temperature. Subsequently, the solutions were
centrifuged at low temperature for 15 min at 12,000 × g. The
supernatants were discarded. Equal volumes of 75% ethanol were
added to the centrifuge tubes and mixed evenly. The solutions were
centrifuged at low temperature for 5 min at 12,000 × g. The
supernatants were discarded, and the precipitates were dissolved in
diethyl pyrocarbonate (DEPC)-treated water.
Reverse transcription
The reagents were added according to the following
proportions, and solutions were mixed evenly. Subsequently, reverse
transcription reaction was started.
10X RT Buffer, 1.5 µl; 10 mM dNTPs, 1.5 µl; RNase
Inhibitor (20 U/µl), 0.2 µl; 5x RT Primer, 3 µl; RNA samples, 5 µl;
Multiscribe™ RT enzyme (50 U/µl), 1 µl; DEPC H2O, 5.5
µl. The thermal profile was as follows: 16°C for 30 min, 42°C for
30 min, 85°C for 5 min, and 4°C for 5 min.
qRT-PCR
The expression of OPG in cells was measured by
RT-PCR. The reaction system was as follows: 2x TaqMan PCR Master
Mix, 10 µl; 10x TaqMan Probe/Primer Mix, 2 µl; cDNA template, 1 µl;
DEPC H2O, 7 µl. The total volume was 20 µl.
The thermal profile was as follows: 94°C for 10 min,
94°C for 20 sec, and 60°C for 1 min, for a total of 40 cycles.
Fluorescence quantitative PCR data were analyzed by the
2−ΔΔCq method.
Total protein extraction. Total cellular protein was
extracted. Cells under healthy growth conditions were washed three
times in phosphate-buffered saline (PBS) to remove floating dead
cells. The cultured cells were then transferred to centrifuge
tubes, and centrifuged for 5 min at 500 × g. Cell precipitates were
then collected. Subsequently, a suitable amount of RIPA buffer was
added, mixed evenly with cells, and placed at 4°C for 20 min.
During mixing, cells were constantly shaken slightly to ensure
complete lysis of cells. The solution was then centrifuged for 20
min at 10,500 × g and 4°C. The supernatants were collected and
stored at −80°C for preservation.
Western blot analysis
The protein content in samples was determined with a
BCA protein quantitative kit according to the manufacturer's
instructions. The final protein concentration was adjusted to 5
µg/µl. Suitable amounts of sample buffer were added to protein
samples obtained by the above steps. Samples were then boiled for 5
min to fully denature proteins. After cooling, samples were loaded
in the wells of gels for SDS-PAGE. A pre-stained marker was used to
accurately determine the size of proteins. The voltage was 100 V
and the separation time was 90 min. When all markers were fully
separated and the bromophenol blue moved to the bottom of gels, the
gels were removed and the marker was carefully identified. The
location of the target protein in the gel was determined according
to the molecular weight of the target protein. A piece of gel
containing the target protein was then cut with a blade. The length
and width of the gel were measured, then two pieces of filter paper
and PVDF membrane of corresponding size were cut and stacked to
prepare a ‘sandwich’. The bubbles between the layers of the
sandwich were expelled using a glass rod. The splint was placed in
the transfer apparatus. Ice cubes and transfer buffer were added to
the tank, the transfer apparatus was placed in an ice bath, and
protein transfer was started. The current was 220 mA, and the time
of transfer was 1.5 h. After transfer, the gel was stained with
Coomassie Brilliant Blue dye to observe protein residues. Membranes
were removed, washed with PBST solution, and a small angle in the
top left corner was cut as a mark on the front and back. Next,
membranes were placed in PBST solution containing 5% skimmed milk
powder and blocked for 1.5 h. The blocking solution was then
discarded. Membranes were placed in a flat plate containing PBST
solution and rinsed 5 times with shaking on the table concentrator
for more than 25 min. Membranes were then placed in clean flat
plates, and the anti-OPG/β-actin primary antibody (1:1,000
dilution) diluted in PBST solution containing 5% skimmed milk
powder was added dropwise to membranes. A layer of preservative
film was used to cover membranes, and they were incubated at 4°C
overnight. After the primary antibody was collected, a suitable
amount of PBST solution was added to flat plates. Plates were
placed on the table concentrator for shock cleaning for 5 min, for
a total of five times. Membranes were placed in clean flat plates,
and the secondary antibody (1:1,000 dilution) diluted in PBST
solution was added dropwise to membranes. A layer of preservative
film was used to cover the membranes, and they were allowed to
incubate at room temperature for 2 h. After the secondary antibody
was removed, a suitable volume of PBST solution was added to the
flat plates. The plates were placed on the table concentrator for
shock cleaning for 5 min, for a total of five times. ECL solution
was added to membranes for signal development. Subsequently, the
membranes were placed in an instrument for acquiring
photographs.
Detecting cell viability by MTT
assay
The wild-type and mutant-type OPG proteins were
purified by affinity chromatography. RAW264.7 cells were
resuspended in α-MEM medium (containing 10% FBS, 2 mmol/l
L-glutamine, 100 IU/ml penicillin, and 100 µg/ml streptomycin), and
seeded in 96-well cell culture plates. Subsequently, the cells were
cultured at 5% CO2, 37°C, and saturated humidity for 24
h. Next, cells continued to be cultured in serum-free α-MEM medium
for 48 h. The wild-type or mutant-type OPG protein at
concentrations of 0, 10, 20, 50 and 100 ng/ml were respectively
added to the appropriate cells. The cells continued to incubate for
24 h. Next, the medium was discarded and cells were washed three
times in PBS. DMEM containing 0.05 mg/ml MTT was added to each
group of cells and they were incubated at 37°C for 4 h. The
supernatant was discarded and 150 µl DMSO was added to each well.
Shock incubation was performed for 10 min. Next, the absorbance
value at 570 nm was measured.
Osteoclast tartrate-resistant acid
phosphatase (TRAP) staining and counting
RAW264.7 cells were re-suspended in α-MEM medium
(containing 10% FBS, 2 mmol/l L-glutamine, 100 IU/ml penicillin,
and 100 µg/ml streptomycin), and seeded in 96-well cell culture
plates. After incubation for 24 h, the medium was replaced with
serum-free α-MEM containing macrophage colony-stimulating factor
(M-CSF) (25 ng/ml) and receptor activator of nuclear factor-κB
ligand (RANKL) (30 ng/ml), followed by incubation for 48 h. The
wild-type or mutant-type OPG proteins at concentrations of 0, 10,
20, 50, and 100 ng/ml were respectively added to the appropriate
cells and were incubated for 3 days. After incubation, TRAP
staining was performed for each group of cells. The amount of
TRAP-positive cells in each group was counted for statistical
analysis.
Detection of bovine cortical bone
slice resorption lacuna
RAW264.7 cells were re-suspended in α-MEM medium
(containing 10% FBS, 2 mmol/l L-glutamine, 100 IU/ml penicillin,
and 100 µg/ml streptomycin), and seeded in 48-well cell culture
plates. After incubation for 24 h, the medium was replaced with
serum-free α-MEM medium containing M-CSF (25 ng/ml) and RANKL (30
ng/ml), and incubated for 48 h. The wild-type or mutant-type OPG
proteins at concentrations of 0, 10, 20, 50 and 100 ng/ml were
respectively added to appropriate cells and incubated for 3 days.
After incubation, bovine cortical bone slices were taken out of the
medium and cleaned by distilled water. Next, bone slice resorption
lacuna were observed and photographed with an XL30-ESEM, and the
bone resorption lacuna area in each group was compared.
Measurement of the expression of genes
related to osteoclast differentiation and activation by RT-PCR
RAW264.7 cells were re-suspended in α-MEM medium
(containing 10% FBS, 2 mmol/l L-glutamine, 100 IU/ml penicillin,
and 100 µg/ml streptomycin), and seeded in 6-well cell culture
plates. After incubation for 24 h, the medium was replaced with
serum-free α-MEM medium containing M-CSF (25 ng/ml) and RANKL (30
ng/ml), and incubated for 48 h. The wild-type or mutant-type OPG
proteins at a concentration of 100 ng/ml were added to the
appropriate cells and incubated for 30 min. After incubation, the
cells were collected and RNA was extracted. Subsequently, the mRNA
levels of the marker genes, TRAP and RANK, under osteoclast
differentiation were measured by RT-PCR.
Statistical analysis
All experiments were repeated three times. Data are
presented as mean ± standard deviation of three independent
experiments. GraphPad 5.0 software (GraphPad Software, Inc., La
Jolla, CA, USA) was used for statistical analyses. ANOVA was used
for comparisons of data. p<0.05 was considered statistically
significant.
Results
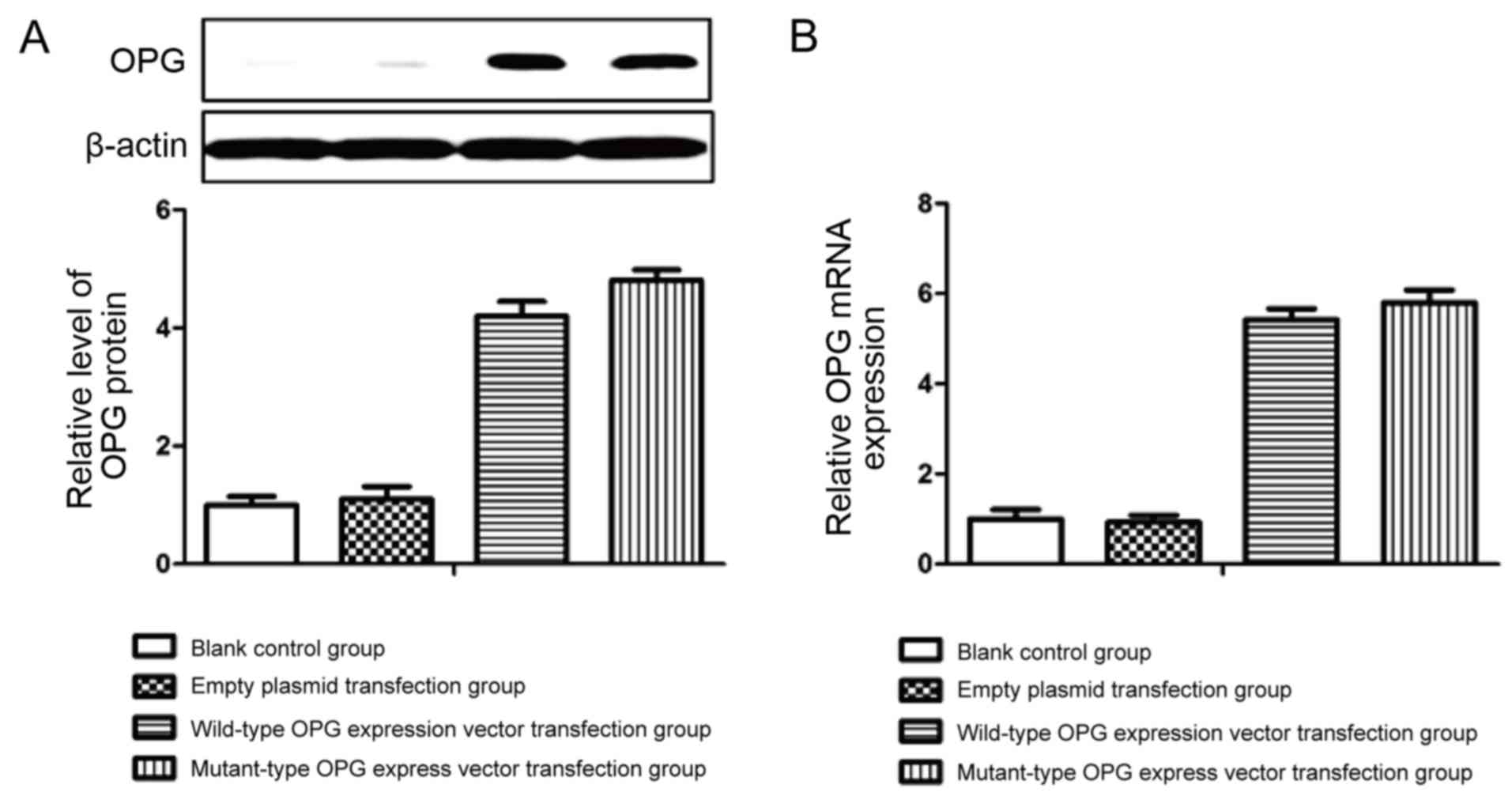
Effect of OPG gene mutation on OPG
mRNA and protein expression
To investigate the effect of the genetic mutation on
OPG mRNA and protein expression, we used site-directed mutagenesis
in vitro to construct the mutant-type OPG expression plasmid
for stable transfection in HEK293 cells. Wild-type and mutant-type
OPG mRNA and protein expression levels were determined by real-time
semi-quantitative PCR and western blot analysis, respectively. As
shown in Fig. 1, the expression of
OPG mRNA and protein in cells transfected with the OPG expression
plasmid was increased. However, the mutation had no effect on OPG
mRNA and protein expression levels in HEK293 cells. Therefore, we
concluded that this mutation did not affect the expression of
OPG.
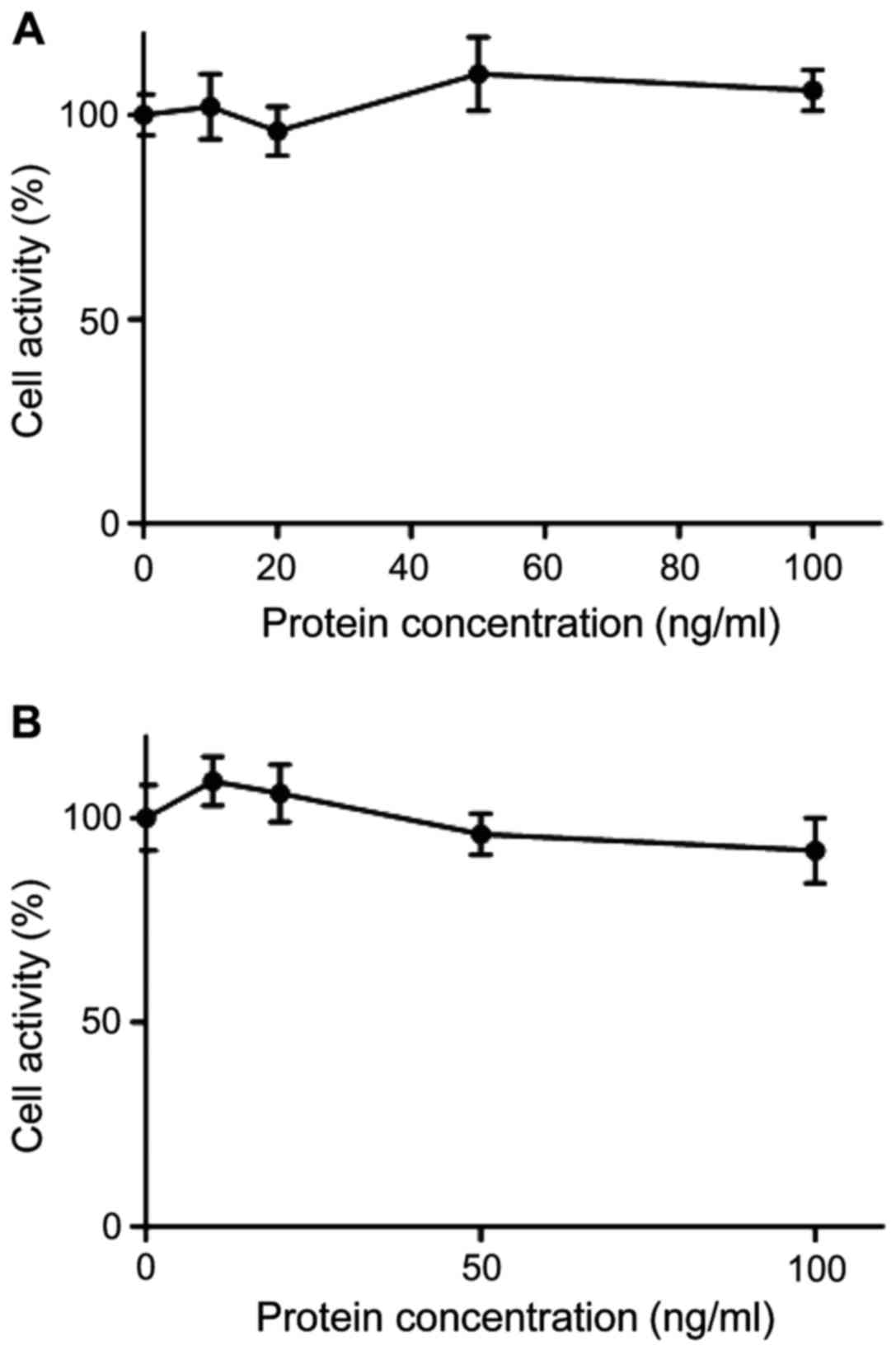
The effect of OPG gene mutation on
cell viability
The wild-type or mutant-type OPG at concentrations
of 0, 10, 20, 50 and 100 ng/ml were respectively added to RAW264.7
cells and incubated for 24 h. Next, the effect of OPG on RAW264.7
cell viability was determined by MTT assay. As shown in Fig. 2, the viability of cells treated with
the wild-type and mutant-type OPG at 100 ng/ml was still over 99%,
which indicated that the wild-type and mutant-type OPG at this
concentration had no cytotoxic effect on RAW264.7 cells.
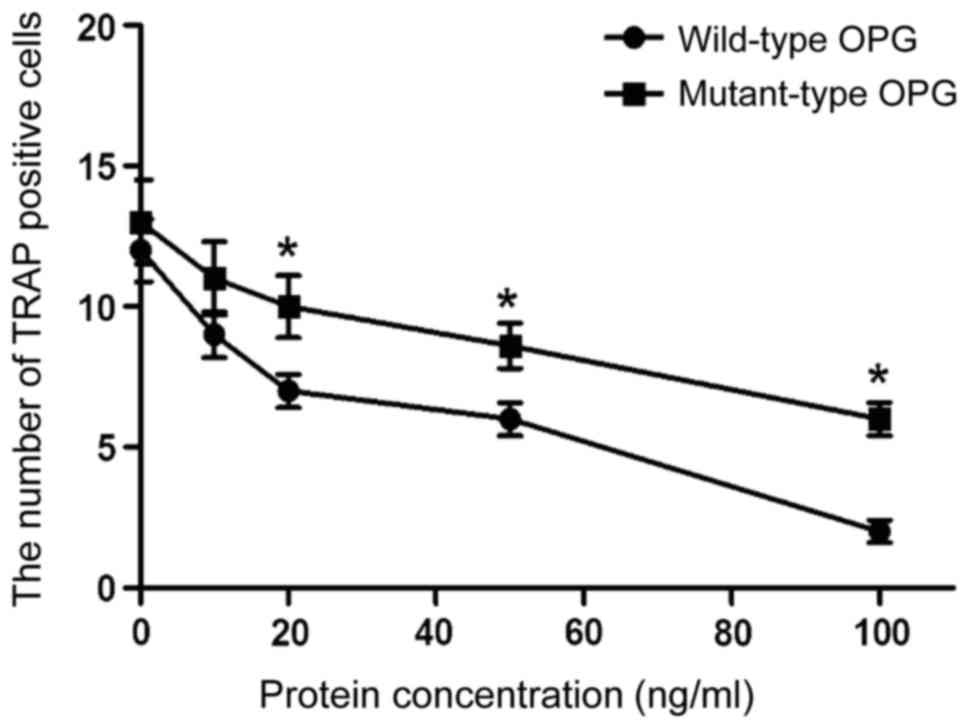
The effect of OPG gene mutation on
osteoclast differentiation
RAW264.7 cells were induced by M-CSF + RANKL, while
different concentrations of wild-type or mutant-type OPG were
respectively added to the appropriate cells. After incubation for 4
days, there were multinuclear macrophages with characteristics of
osteoclasts, such as being positive for TRAP. As shown in Fig. 3, the number of TRAP-positive cells
decreased with increasing concentration of wild-type or mutant-type
OPG. At the concentrations of 20, 50 and 100 ng/ml, the inhibitory
effect of wild-type OPG was significantly higher than that of
mutant-type OPG (p<0.05). These results demonstrate that the
mutant-type OPG can affect the differentiation of osteoclasts.
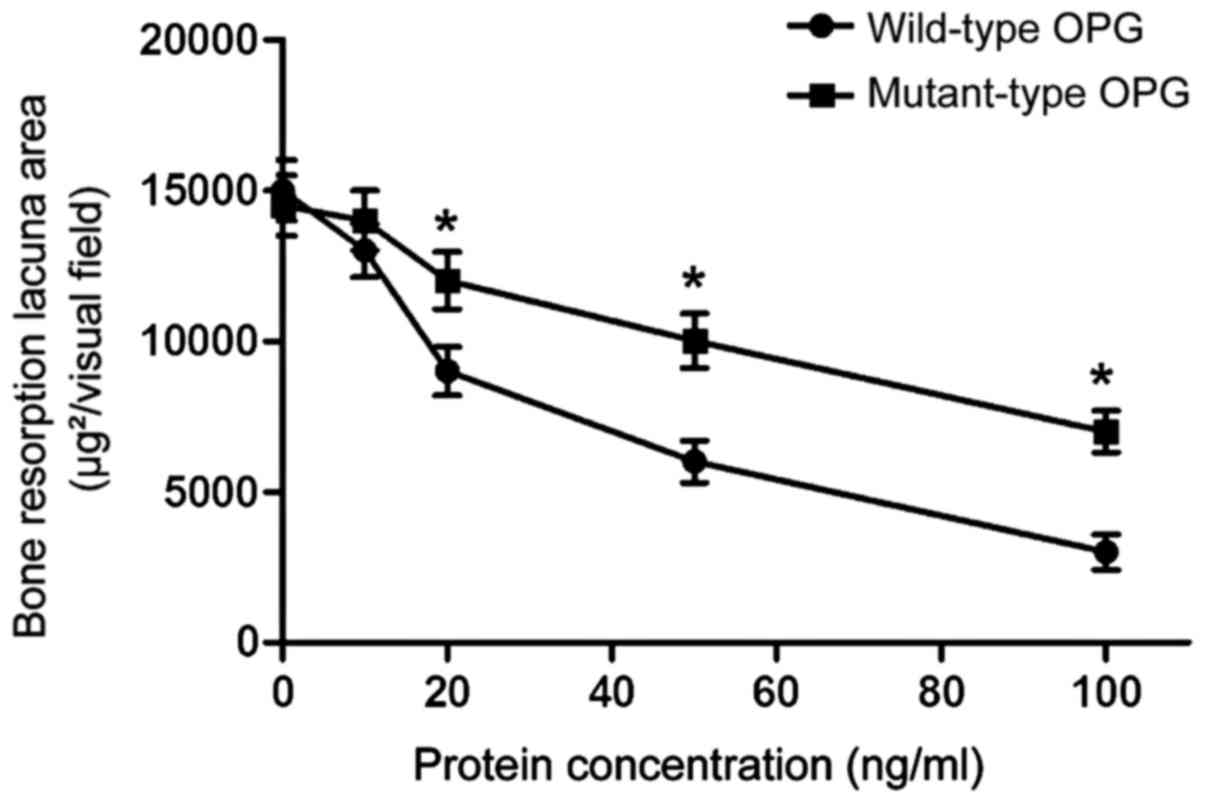
The effect of OPG gene mutation on
bone resorption ability of osteoclasts
RAW264.7 cells were induced by M-CSF + RANKL and
differentiated into mature osteoclasts with bone resorption
activity. Different concentrations of wild-type or mutant-type OPG
were added respectively to each group. Various shapes of absorption
lacuna appeared on bovine cortical bone slices. The smaller the
area of resorption lacuna, the lower the bone resorption activity
was. As shown in Fig. 4, both
mutant-type and wild-type OPG inhibited the bone resorption
activity of osteoclasts. The inhibitory effect of wild-type OPG was
significantly higher than that of mutant-type OPG at the
concentrations of 20, 50 and 100 ng/ml (p<0.05). These results
indicate that mutant-type OPG can affect the bone resorption
ability of osteoclasts.
The effect of OPG gene mutation on
expression of genes related to osteoclast differentiation
RAW264.7 cells were treated with 100 ng/ml wild-type
or mutant-type OPG. Next, the mRNA levels of the marker genes, TRAP
and RANK, during the process of osteoclast differentiation were
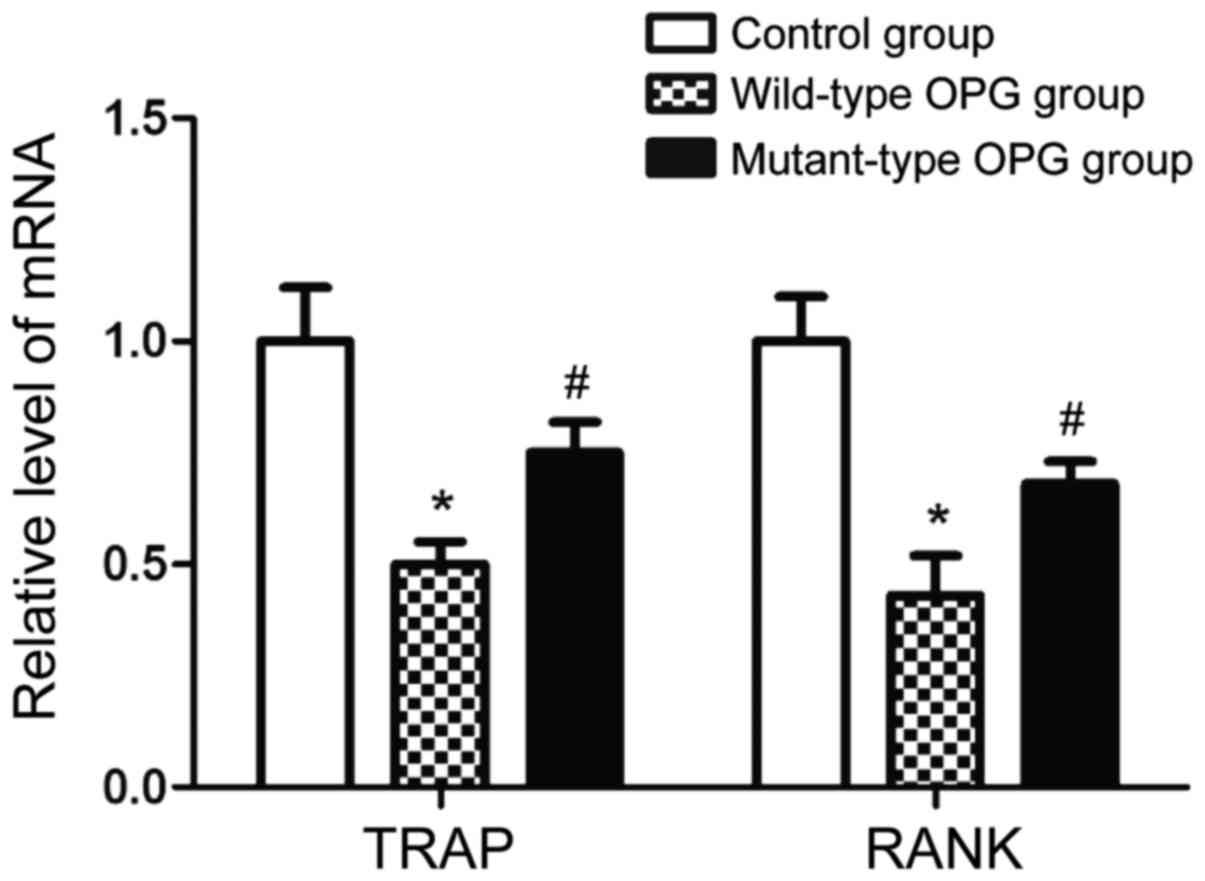
measured by RT-PCR. As shown in Fig.
5, the levels of TRAP and RANK mRNA in the wild-type OPG
treatment group were significantly lower than those in the control
group, while the levels of TRAP and RANK mRNA in the mutant-type
OPG treatment group were significantly lower than those in the
wild-type OPG treatment group (p<0.05). These results indicate
that OPG gene mutation can affect osteoclast differentiation.
Discussion
Osteoporosis is a systemic bone disease that is
characterized by decreased osteopenia and degeneration of the
microstructure of bone tissues, thereby increasing bone fragility
and the risk of fracture (6). Bone
formation and bone resorption are the two basic processes of bone
metabolism, and they are maintained in a dynamic balance. New bone
is constantly being generated, while old bone is constantly being
absorbed. Osteoclasts, which are a component of bone tissues,
function in bone resorption. They exert a synergistic action with
osteoblasts, which jointly play an important role in the process of
bone development and formation. Both pathological increases and
decreases of osteoclast activity can cause diseases, such as
osteoporosis, bone sclerosis, and Paget disease. As a secreted
glycoprotein, OPG is a member of the TNF receptor family, which
exists as two forms, the monomer and homodimer (7,8). A
previous study showed that OPG gene knockout mice aged 1–2 months
manifested severe osteoporosis and had complete absence of bone
trabecula (9). Furthermore, the long
bone, vertebra, and pelvis of transgenic mice with overexpression
of OPG manifested obvious symptoms of bone sclerosis (10). We identified the genetic
polymorphism, g.27563G>A, within the fifth exon of the OPG gene
by CRS-PCR. The female BMD associated with the GG genotype was
significantly higher than that of the GA and AA genotypes. However,
the specific association between OPG gene polymorphisms and
osteoporosis remains unclear. Based on the successful establishment
of the cell culture system with overexpression of the wild-type and
mutant-type OPG gene, this study aimed to investigate the effect of
the OPG gene mutation on its protein expression. The results
indicated that the OPG gene mutation had no significant effect on
its protein expression levels.
Osteoclast activation is the basis of bone
resorption, and involves the transformation of mature osteoclasts
in a resting state to an active state. This includes migration and
adhesion to mineralized bone matrix, formation of the bone
resorption microenvironment, and synthesis and secretion of a
variety of functional enzymes. M-CSF and RANKL are two
indispensable factors for osteoclast differentiation and
activation. Under the combined action of M-CSF and RANKL,
osteoclast precursors initially fuse to become osteoclasts, which
are characterized by TRAP-positivity until they mature. Only by
activation can mature osteoclasts acquire bone resorption activity.
Hofbauer et al reported that OPG can regulate osteoclast
differentiation, activation, and survival through the
OPG-RANKL-RANK axis, thereby affecting skeletal metabolism in the
body (11,12). Therefore, whether the proteins
encoded by the wild-type and mutant-type OPG genes act differently
during differentiation and activation of osteoclasts requires
further investigation. The results of this study showed that
wild-type OPG inhibited the differentiation of osteoclasts, while
the inhibitory effect of mutant-type OPG was significantly lower
than that of the wild-type protein. However, the specific mechanism
of action remains unclear and further research is needed.
In conclusion, based on the successful establishment
of a cell culture system with overexpression of the wild-type and
mutant-type OPG gene, this study aimed to investigate the effect of
an OPG gene mutation on its protein expression and activity. The
results indicated that the genetic mutation did not affect the
protein expression levels of OPG, but inhibited the normal activity
of the OPG gene.
References
|
1
|
McCormick RK: Osteoporosis: Integrating
biomarkers and other diagnostic correlates into the management of
bone fragility. Altern Med Rev. 12:113–145. 2007.PubMed/NCBI
|
|
2
|
Riasnyĭ VM, Apukhovs'ka LI, Velykyĭ MM,
Shymans'kyĭ IO, Labudzyns'kyĭ DO and Komisarenko SV:
Immunomodulatory effects of vitamin D3 and bisphosphonates in
nutritional osteoporosis in rats. Ukr Biokhim Zh (1999). 84:73–80.
2012.(In Ukrainian). PubMed/NCBI
|
|
3
|
Hwang JS, Chen JF, Yang TS, Wu DJ, Tsai
KS, Ho C, Wu CH, Su SL, Wang CJ and Tu ST: The effects of strontium
ranelate in Asian women with postmenopausal osteoporosis. Calcif
Tissue Int. 83:308–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hofbauer LC and Schoppet M:
Osteoprotegerin gene polymorphism and the risk of osteoporosis and
vascular disease. J Clin Endocrinol Metab. 87:4078–4079. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohmori H, Makita Y, Funamizu M, Hirooka K,
Hosoi T, Orimo H, Suzuki T, Ikari K, Nakajima T, Inoue I, et al:
Linkage and association analyses of the osteoprotegerin gene locus
with human osteoporosis. J Hum Genet. 47:400–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dominguez LJ, Scalisi R and Barbagallo M:
Therapeutic options in osteoporosis. Acta Biomed. 81 Suppl 1:55–65.
2010.PubMed/NCBI
|
|
7
|
Montagnana M, Lippi G, Danese E and Guidi
GC: The role of osteoprotegerin in cardiovascular disease. Ann Med.
45:254–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brosch S, Redlich K and Pietschmann P:
Pathogenesis of osteoporosis in rheumatoid arthritis. Acta Med
Austriaca. 30:1–5. 2003.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yasuda H, Shima N, Nakagawa N, Mochizuki
SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, et
al: Identity of osteoclastogenesis inhibitory factor (OCIF) and
osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits
osteoclastogenesis in vitro. Endocrinology. 139:1329–1337. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wright HL, McCarthy HS, Middleton J and
Marshall MJ: RANK, RANKL and osteoprotegerin in bone biology and
disease. Curr Rev Musculoskelet Med. 2:56–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hofbauer LC, Kühne CA and Viereck V: The
OPG/RANKL/RANK system in metabolic bone diseases. J Musculoskelet
Neuronal Interact. 4:268–275. 2004.PubMed/NCBI
|
|
12
|
Khosla S: Minireview: The OPG/RANKL/RANK
system. Endocrinology. 142:5050–5055. 2001. View Article : Google Scholar : PubMed/NCBI
|