Introduction
Measles is a highly infectious and potentially
dangerous disease, and a major killer of children (1). Subsequent to contact with MV, almost
all children without immunity are infected. The clinical
characteristics of measles include fever, upper respiratory tract
infection, conjunctivitis, oral cavity mucous membrane measles,
maculopapule on the whole body and pigmentation after back rash
(2). MV, a paramyxovirus of the
Morbillivirus genus of the paramyxovirus family, is an
enveloped virus containing a single-stranded, minus (−) sense 50S
RNA genome (3). MV infection causes
profound immunosuppression, which makes measles patients
susceptible to secondary infections accounting for high morbidity
(4). It is responsible for an acute
childhood illness that infects >40 million individuals and leads
to >1 million mortalities per annum (5). It is generally thought that patients
with measles have infectivity 5 days prior to and after its
outbreak and the infective period may be prolonged to 10 days when
complications are present.
Genetic heterogeneity exists in the wild-type MV,
but there is only one serotype (6).
Since the 1980s, MV has had a genetic drift in all countries, as
indicated by the antigenic variation of MV and nucleotide sequence
analysis. The MV P gene encodes eight proteins, including the P
protein and the two non-structural proteins C and V (7). The proteins C and V are formed by
selective translation of viral RNA encoding phosphoprotein. For the
other 6 structural proteins, P protein, giant protein (L protein)
and nucleocapsid protein (N protein) constitute a capsid wrapping
viral RNA, while hemagglutinin protein (H protein), fusion protein
(F protein) and matrix protein (M protein) combine with lipids from
the membrane of host cells to form the envelope of virus (8,9). The N
protein of MV consists of an N-terminal moiety, N CORE, which is
resistant to proteolysis and a C-terminal moiety, N TAIL, which is
hypersensitive to proteolysis and not visible as a distinct domain
by electron microscopy (10). At
present, MV is thought to have 23 genotypes, including A,
B1-B3, C1 and C2,
D1-D10, E, F, G1-G3 as
well as H1 and H2, 5 of which (B1,
D1, E, F and G1) have no activity since their
genotypes have not been found in the past 15 years (11). Gene sequence analysis of MV isolated
from Chinese provinces including Shandong, Hebei, Hunan, Beijing,
Anhui, Hainan and Shanxi showed that the H1 genotype is
predominant in China and that intra-type variation will continue
(12,13).
Various MV genotypes have a differential
geographical distribution and various regions have different local
epidemic strains (11,13–15). In
addition, time has a certain relevance in the global epidemic of
measles, providing a theoretical basis for monitoring the global
molecular epidemiology of measles. Yunnan is a Chinese province
with a high incidence of measles due to its unique geographical
location, bordering with certain Southeast Asian countries with a
high incidence of measles (15). At
present, the clinical manifestations of measles in pediatric
patients are diverse with heterogeneity in symptoms and occurrence
of complications. In addition, to the best of our knowledge, no
previous study has assessed the molecular epidemiology of MV in
Kunming (China).
The present study assessed the molecular
epidemiology of MV in 38 pediatric patients with measles in Kunming
from 2008 by reverse-transcription quantitative polymerase chain
reaction (RT-qPCR) analysis to determine the major genotype of
wild-type MV in Kunming. The degree of genotype variation and the
possible association with differential clinical manifestations
between patients were also assessed.
Materials and methods
Sample collection and preparation
The study was approved by Ethics Committee of the
Second Affiliated Hospital of Kunming Medical University (Kunming,
China). The parents or guardians of all patients provided written
informed consent prior to enrollment in the study.
Pediatric patients (age, <12 years) were
recruited at the Second Affiliated Hospital of Kunming Medical
University (Kunming, China) and the Children's Hospital of Kunming
City (Kunming, China) between January 1, 2008 and March 31, 2008,
and 38 cases (21 from the Second Affiliated Hospital of Kunming
Medical University and 17 from the Children's Hospital of Kunming
City) were selected. Venous blood (2 ml) was collected from each
patient and urine was collected from 30 of the 38 cases. The
inclusion criteria conformed to the diagnostic criteria for measles
included in the seventh edition ‘pediatrics’ (16) and the disease being in its infectious
stage (3 days before or 6 days after rash; this duration was chosen
by researchers to collect positive samples).
The samples were stored at −80°C. Blood samples were
centrifuged at 4°C, 5,478.2 × g for 10 min to obtain serum samples,
which were stored at −20°C until analysis.
RNA isolation and RT-PCR
For RNA isolation, total MV RNA was extracted from
urine specimens using the mirVana miRNA isolation kit (Ambion;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) after the sample
collection. After isolation, the RNA concentration was assessed
using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific,
Inc.) and the RNA solution was stored at −80°C for further use.
RT-PCR was performed in 50-µl reaction volumes. The
amplification reaction mixture contained 0.2 mM of each primer, 1X
Ex-Taq buffer, 0.25 mM of each deoxynucleotide phosphate, 0.25 U
Ex-Taq DNA polymerase (Takara Bio Inc., Otsu, Japan) and 2.5 µl of
the extracted template DNA in DNAse-free water. The upstream primer
of measles virus was 5′-GCTATGCCATGGGAGTAGGAGTGG−3′, and the
downstream primer was 5′-CCTCGGCCTCTCGCACCTAGT-3′. Sequence data
were deposited in GenBank under accession number (EU090820). The
reaction conditions were as follows: Initially denaturation at 95°C
for 5 min, followed by 35 cycles of denaturation at 95°C for 30
sec, annealing at 52°C for 30 sec and elongation at 72°C for 1 min,
and a final elongation step at 72°C for 7 min. Distilled water was
used as a negative control here.
The amplified products (50 µl) were visualized by
staining with ethidium bromide and separation by electrophoresis on
1.0% agarose gels (Beijing Borunlaite Science & Technology Co.,
Ltd., Beijing, China). The PCR products were purified with a
BioSpinGel Extraction kit (Bioer Technology Co., Ltd., Beijing,
China). The PCR products were purified and cloned into the pEASY-T1
vector (Beijing TransGen Biotech Co., Ltd., Beijing, China)
(17). The vectors were then
transduced into Trans1-T1 Phage Resistant Chemically Competent
Cells (Beijing TransGen Biotech Co., Ltd.). The positive clones
were selected and sequenced by Invitrogen Shanghai (Thermo Fisher
Scientific, Inc.). The sequencing results were edited using DNAStar
software 7.0 (DNAStar, Inc., Madison, WI, USA).
Detection of antibodies of MV
A total of 38 serum samples were evaluated for
antibodies using ELISA. The procedures for detection were also
performed according to the manufacturer's instructions.
Sequence analysis
The nucleotide sequences designated in the present
study were compared with the 23 genotypes of MV by using the Basic
Local Alignment Search Tool (Blast; https://blast.ncbi.nlm.nih.gov/Blast.cgi). Relevant
sequences were also downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). The sequences
were assembled and aligned using DNAStar (http://www.dnastar.com/). To analyze the association
between the variation of MV and clinical manifestation of measles
in infected patients, a tree was generated based on the sequences
of the complete H1/2 gene. Phylogenetic analysis was
performed using the MP method in the software MEGA 5.0 (http://www.megasoftware.net/) and the European
Bioinformatics Institute Tool ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/), and the
confidence level of the branch was assessed using bootstrap
analysis with 1,000 replicates. The tree was rooted, and human
parvovirus B19 and MV were used as out groups.
Statistical analysis
Differences between two proportions were measured by
using Pearson's Chi-squared test or Fisher's exact test. P<0.05
was considered to indicate a statistically significant difference.
Statistical analyses were performed by using SPSS 13.0.0
statistical software for Windows (SPSS, Inc., Chicago, IL,
USA).
Results
MV determination
Urine specimens were collected from 30 of the 38
pediatric patients and 7 strains were isolated with a positive rate
of 23.33%. The results demonstrated that it was possible to isolate
MV from urine specimens of patients up to 6 days after presenting
with a rash. The positive rate for the 21 patients whose specimens
were taken 1–3 days after rash was 19.05% and that of the 9
patients whose samples were taken at 4–6 days after the rash was
33.33%. Statistical analysis of the positive rate for the two
time-periods using Fisher's exact test revealed no statistically
significant difference (P=0.640). Venous blood was collected from
38 patients with measles and serum antibody IgM was determined. The
results showed that the positive rate of IgM antibody was 100%.
Statistical analysis of the positive rate of MV in urine specimens
compared with that of serum IgM antibody revealed a statistically
significant difference (χ2=44.024; P<0.001).
RT-PCR of N gene
Seven strains of MV isolated from urine specimens of
30 subjects were amplified and purified. After identification by
1.5% agarose gel electrophoresis, an obvious positive band was
observed at 516 bp (Fig. 1). Four
strains were selected from the 7 strains of MV after purification.
Sequence determination analysis was automatically completed on an
ABI 3730 sequencer by Invitrogen Shanghai (Thermo Fisher
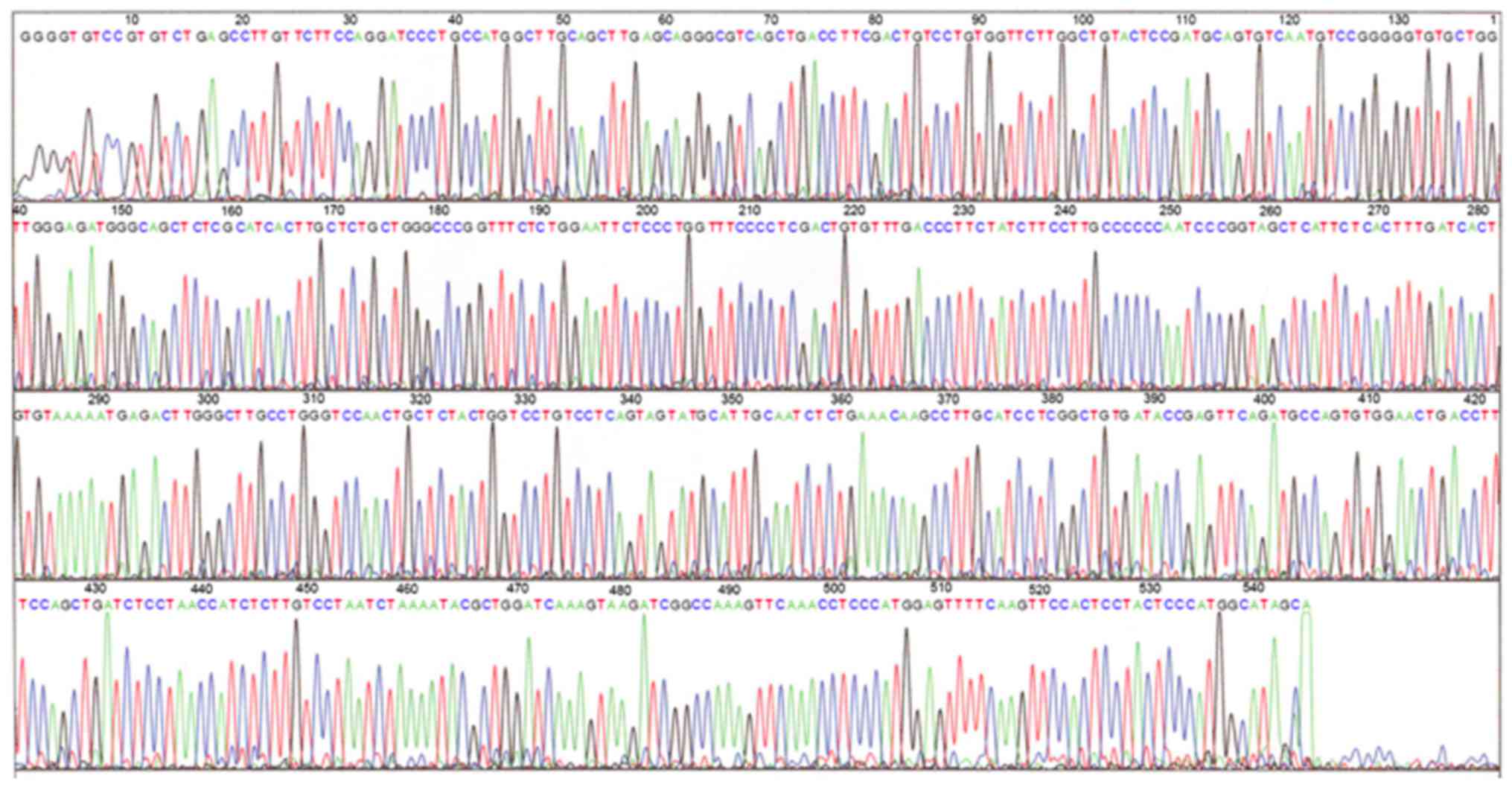
Scientific, Inc.) and representative sequencing data are presented
in Fig. 2.
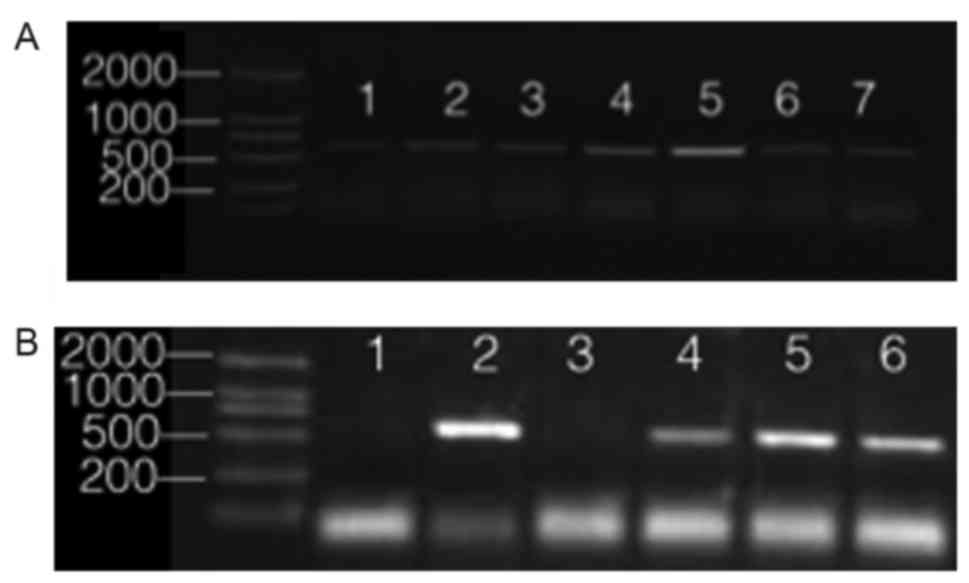 | Figure 1.Results of agarose gel electrophoresis
of MV. (A) Nucleotide fragment amplification of N gene in MV.
Lane1, Kunming08-6 strain; lane 2, Kunming08-1 strain; lane 3,
Kunming08-5 strain; lane 4, Kunming08-2 strain; lane 5, Kunming08-3
strain; lane 6, Kunming08-7 strain; lane 7, Kunming08-4 strain. (B)
Electrophoresis chart of the 4 MV strains after purification. MV,
measles virus. Lane 1, ddH2O negative control; lane 2,
Kunming08-3 strain; lane 3, ddH2O negative control; lane
4, Kunming08-1 strain; lane 4, Kunming08-1 strain; lane 5,
Kunming08-2 strain; lane 6, Kunming08-3 strain. |
Sequence analysis
Analysis of genetic kinship
A total of 543 nucleotide sequences at the carboxyl
terminal of the N gene in 4 strains of wild-type MV were compared
with the 23 genotypes of MV from GenBank by using the Basic Local
Alignment Search Tool (Blast; https://blast.ncbi.nlm.nih.gov/Blast.cgi). The results
demonstrated that all of the 4 strains of MV were of the H
genotype, which was consistent with the predominant genotype in
China. MEGA 5.0 software and the European Bioinformatics Institute
Tool ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) were then
used to perform a genetic kinship analysis between the 543
nucleotide sequences at the carboxyl terminal of the N gene in the
4 strains of wild-type MV and the representative strain at WHO for
China between 1993 and 1994 and the corresponding sequences of
referential strains (China93-7, China93-2, China94-7 and China94-1)
for the H1 and H2 genotypes (13). The genetic kinship trees of
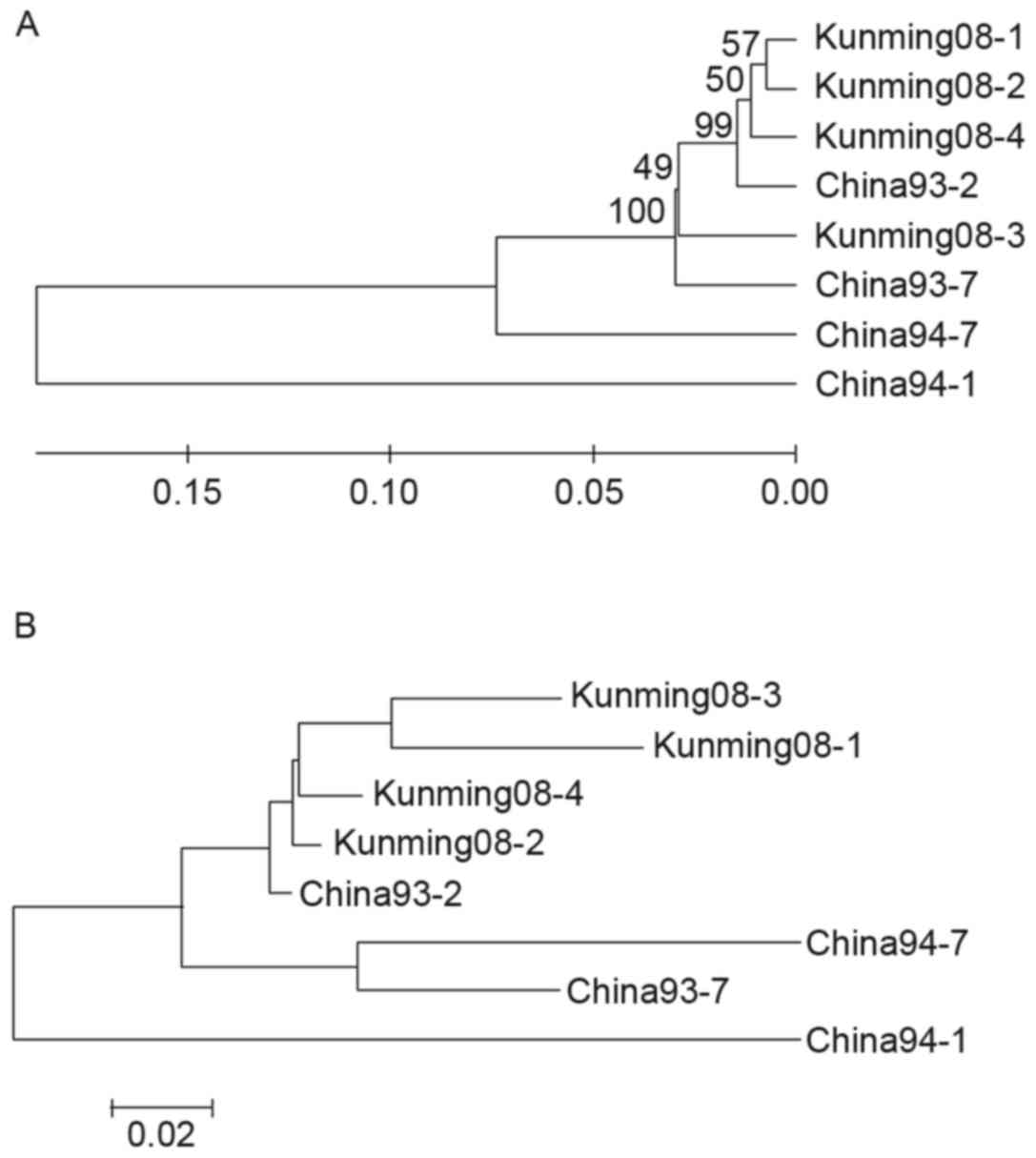
nucleotide and amino acid sequences are displayed in Fig. 3A and B, respectively. The 4 strains
of MV were all of the H1 genotype and belonged to the
same branch as China93-7. The kinship between isolated MV and
referential strain China93-2 (H1a subtype) was closest.
It was therefore indicated that the genotype of the 4 MV strains
was the H1a subtype of the H1 genotype.
Variation analysis of nucleotides and amino
acids
Genetic distance analysis within and between gene
sequences of 4 MV strains and WTO referential strains China93-7,
China93-2, China94-7 and China94-1 were performed. The results
presented in Table I demonstrated
that the genetic distance between nucleotide sequences of the 4 MV
strains from the H1 and H2 genotype
referential strains China93-7 and China94-1 was 0.031–0.047 and
0.332–0.398, respectively. The genetic distance for the
H1a genotype (standard strain China93-2) was
0.010–0.033, and that between the 4 strains was 0.005–0.040.
 | Table I.Genetic distance analysis of the
nucleotides and amino acids of the 4 wild-type measles virus
strains isolated from Kunming area and referential strains. |
Table I.
Genetic distance analysis of the
nucleotides and amino acids of the 4 wild-type measles virus
strains isolated from Kunming area and referential strains.
| A, Nucleotides |
|---|
|
|---|
| Strain | Kunming08-1 | Kunming08-2 | Kunming08-3 | Kunming08-4 | China93-7 | China93-2 | China94-7 | China94-1 |
|---|
| Kunming08-1 |
|
|
|
|
|
|
|
|
| Kunming08-2 | 0.005 |
|
|
|
|
|
|
|
| Kunming08-3 | 0.029 | 0.039 |
|
|
|
|
|
|
| Kunming08-4 | 0.016 | 0.008 | 0.040 |
|
|
|
|
|
| China93-7 | 0.031 | 0.035 | 0.047 | 0.047 |
|
|
|
|
| China93-2 | 0.010 | 0.018 | 0.033 | 0.029 | 0.039 |
|
|
|
| China94-7 | 0.114 | 0.107 | 0.167 | 0.089 | 0.160 | 0.132 |
|
|
| China94-1 | 0.353 | 0.348 | 0.398 | 0.332 | 0.399 | 0.380 | 0.271 |
|
|
| B, Amino acids |
|
| Strain | Kunming08-1 | Kunming08-2 | Kunming08-3 | Kunming08-4 | China93-7 | China93-2 | China94-7 | China94-1 |
|
| Kunming08-1 |
|
|
|
|
|
|
|
|
| Kunming08-2 | 0.010 |
|
|
|
|
|
|
|
| Kunming08-3 | 0.059 | 0.042 |
|
|
|
|
|
|
| Kunming08-4 | 0.003 | 0.010 | 0.052 |
|
|
|
|
|
| China93-7 | 0.066 | 0.073 | 0.073 | 0.059 |
|
|
|
|
| China93-2 | 0.156 | 0.156 | 0.156 | 0.149 | 0.197 |
|
|
|
| China94-7 | 0.188 | 0.213 | 0.273 | 0.205 | 0.282 | 0.282 |
|
|
| China94-1 | 0.088 | 0.095 | 0.118 | 0.088 | 0.149 | 0.118 | 0.273 |
|
The genetic distance between amino acid sequences of
the 4 MV strains and those of the H1 and H2
genotype referential strains China93-7 and China94-1 was
0.088–0.149 and 0.205–0.282, respectively. The distance for the H1a
genotype was 0.003–0.066, and that between the 4 strains was
0.010–0.073.
Homology analysis of nucleotides and amino
acids
The homology of 543 nucleotides at the carboxyl
terminal of the N gene in the 4 strains of wild-type MV was
96–98.9%. The nucleotides differed in 6–12 bases and the homology
of amino acids was 95.6–99.6%. The nucleotide homology of the 4
strains of wild-type MV with the H1 standard strain
China93-7 was 95.9–99.7%, while the amino acid homology was
89.9–94.8%. The nucleotide homology with the H1a standard strain
China93-2 was 96.9–99.8% and the amino acid homology was 94.8–99.7%
(Table II).
| Table II.Homology analysis of nucleotides and
amino acids of the 4 wild-type measles virus strains isolated from
Kunming area and referential strains. |
Table II.
Homology analysis of nucleotides and
amino acids of the 4 wild-type measles virus strains isolated from
Kunming area and referential strains.
| A, Nucleotides |
|---|
|
|---|
| Strain | Kunming08-1 | Kunming08-2 | Kunming08-3 | Kunming08-4 | China93-7 | China93-2 | China94-7 | China94-1 |
|---|
| Kunming08-1 |
|
|
|
|
|
|
|
|
| Kunming08-2 | 0.969 |
|
|
|
|
|
|
|
| Kunming08-3 | 0.960 | 0.969 |
|
|
|
|
|
|
| Kunming08-4 | 0.978 | 0.989 | 0.978 |
|
|
|
|
|
| China93-7 | 0.959 | 0.988 | 0.959 | 0.997 |
|
|
|
|
| China93-2 | 0.969 | 0.998 | 0.969 | 0.988 | 0.989 |
|
|
|
| China94-7 | 0.869 | 0.886 | 0.868 | 0.897 | 0.915 | 0.906 |
|
|
| China94-1 | 0.778 | 0.787 | 0.778 | 0.808 | 0.819 | 0.809 | 0.859 |
|
|
| B, Amino acids |
|
| Strain | Kunming08-1 | Kunming08-2 | Kunming08-3 | Kunming08-4 | China93-7 | China93-2 | China94-7 | China94-1 |
|
| Kunming08-1 |
|
|
|
|
|
|
|
|
| Kunming08-2 | 0.959 |
|
|
|
|
|
|
|
| Kunming08-3 | 0.956 | 0.968 |
|
|
|
|
|
|
| Kunming08-4 | 0.956 | 0.996 | 0.969 |
|
|
|
|
|
| China93-7 | 0.899 | 0.927 | 0.909 | 0.948 |
|
|
|
|
| China93-2 | 0.948 | 0.996 | 0.959 | 0.997 | 0.929 |
|
|
|
| China94-7 | 0.839 | 0.877 | 0.868 | 0.868 | 0.906 | 0.868 |
|
|
| China94-1 | 0.789 | 0.858 | 0.788 | 0.827 | 0.789 | 0.855 | 0.788 |
|
Epidemiology analysis
Morbidity situation
The number of pediatric patients who presented with
measles at the Children's Hospital of Kunming City and the Second
Affiliated Hospital of Kunming Medical University between January
2008 and March 2008 was 51, while that in January, February and
March 2006 was 122, 113 and 129, respectively. The numbers in
January, February and March of 2007 were 124, 55 and 35,
respectively. The morbidity decreased with increasing time, which
may be associated with the strength of immunization to MV in 2006
due to a pandemic of measles.
Age distribution
The age of the 38 pediatric patients with measles
enrolled in the present study ranged from 51 days to 11 years. The
number of children below the age of 1 year was 21 and accounted for
55.26% of the total patients selected. The number of children aged
from 1 to 2 years old was 9, accounting for 23.68%. The number of
patients aged between 2–3, 7–8 and 8–9 years old was 2 in each age
range, respectively, accounting for 5.26% of all the recruited
children. In total, the number of patients below the age of 2 years
was 30 and accounted for 78.95% of the 38 patients. A high
incidence of measles was therefore encountered in children aged
<2 years. Within the cohort of 38 pediatric patients, there were
23 male and 15 females with a proportion of 60.53 and 39.47%,
respectively, indicating that the amount of male children was much
higher than that of female children.
Regional distribution
Within the cohort of the present study (n=38), the
regional distribution within Kunming was as follows: Wuhua, 7;
Panlong, 4; Guandu, 5; Xishan, 7; Dongchuan, 1; Chenggong, 1;
Jinning, 2; Fumin, 2; Chongming, 1; Luquan, 7; and Anning, 1. This
indicated that the number of patients with measles in Wuhua and
Xishan was highest in Kunming city. The cases of measles in the
present study were sporadic cases and not affected by any pandemic.
The differences in the number of infected patients in different
regions may be associated with the imbalance of immunization work.
Within the cohort comprising 38 cases, 7 had been vaccinated
against measles, 30 had not been vaccinated and the vaccination
status of the remaining case was unknown. Cases with no immunity
and ominous immune history accounted for 81.58% of the total
patient population. Among the 7 patients vaccinated against
measles, 5 had primary immunization failure and the other 2, who
were aged >7 years, had received no second vaccination.
Association between clinical manifestation and
genotype
Four strains were randomly selected from the 7
strains of MV isolated from urine specimens of measles patients.
Sequence determination and analysis showed that they were all of
the H1a genotype, but there was a great difference the
clinical manifestation of measles in the 4 patients (Table II). This indicated that the clinical
manifestation of measles in different patients was not obviously
associated with the genotype of MV.
Discussion
Measles is an acute viral infectious disease caused
by MV and is characterized by fever, upper respiratory tract
infection and maculopapules over the entire body surface (18). According to an estimation by the
world health organization, ~45 million measles cases occur and 1
million children die from measles and its complications each year
(19). Monitoring of the genotype
distribution of MV in different areas is of important significance
for identifying scientific approaches for blocking the spread of MV
and developing a regional measles elimination plan (20).
MV is a RNA virus of the genus Morbillivirus
within the family Paramyxoviridae, and each virus has antigen
cross-reactivity within the Morbillivirus genera (21). MV only has one serotype, but it has
multiple genotypes. In the present study, 38 blood samples and 30
urine specimens were collected from patients with measles. Previous
studies showed that at the prodromal stage of measles and on the
first day that a rash appears on the body, MV is easy to be
separated from the patient's blood and respiratory secretions
(22). In addition, studies reported
that multinuclear giant cells in the nasopharynx of measles
patients were rapidly reduced after the rash, but multinuclear
giant cells were still found in the urine sediment a few days later
(23). Therefore, the time window
for virus collection from the urine may be longer than that from
other fluids. The results of the present study showed that MV may
be isolated from urine specimens within 6 days after rash. The
virus separation rate within 4–6 days after rash (33.33%) was
higher than that within 3 days (19.05%), while there was no
statistical difference between them. In addition, the determination
of measles IgM antibody from the serum of the 38 cases of measles
at the acute stage provided a positive rate of 100%. This indicated
that the detection rate of measles IgM antibody from serum was
evidently higher than the separation rate of MV from urine.
The H and N genes have the greatest variation among
the structural genes of MV (24).
The N gene encodes 525 amino acids, the H gene encodes 617 amino
acids and they have 7% nucleotide variation in their sequences
(15). Particularly the 450
nucleotides at the carbon end of the N gene is the largest zone for
MV gene variations and the degree of genetic variation of MV is up
to 12%. The 1,851 nucleotides contained in the H gene, 1,575 in the
N gene and 450 in the N gene carbon end were the main subjects of a
previous epidemiological study on MV (25). Their sequence analysis results have
been widely used to describe the genotypes of MV. In the present
study, 4 MV strains were randomly selected from the 7 isolated MV
strains and a molecular epidemiological analysis was performed.
Blast comparison and genetic affinity analysis of the 543
nucleotides at the carboxyl terminal of the N gene in 4 strains of
wild-type MV and 23 genotypes of MV from GenBank as well as a
vaccine strain was performed. A total of 4/23 genotypes were
Chinese representatives, including China93-7, China93-2, China94-7
and China94-1. By comparison with these 4 strains which were the
most common genotypes in China, genetic drift and variation would
be confirmed and this would help to build the surveillance of MV
and to evaluate the efficacy of vaccination. The results showed
that the sequences from all of the 4 strains of MV were of the H
genome and of the H1a subtype. The genotype of the 4
wild-type MV strains from Kunming area was consistent with the
genotype of the major epidemic strain in China. The genetic
distance, nucleotide homology and amino acid homology between the
corresponding sequences of the 543 nucleotides at the carboxyl
terminal of the N gene of the 4 wild-type MV strains and
referential strains were assessed. The results revealed only a
minor difference between the 4 MV strains. The genotype of the
wild-type MV strains encountered in Kunming area from January 2008
to March 2008 was consistent with that reported in Yunnan province
in 2005 (26).
Yunnan province has planned immunization-projects
since the early 1980s and measles has been effectively controlled
due to the enhancement of the measles vaccine inoculation rate
(27). Kunming is the capital of
Yunnan province and the epidemiological characteristics of measles
in Kunming area have a certain representativeness. The present
study assessed the age at onset and gender distribution,
epidemiology of measles as well as genetic variations of MV and
their association with clinical manifestation of measles in
patients. The results demonstrated that the clinical manifestation
of measles had no obvious correlation with the genotype of
wild-type MV.
In conclusion, the preponderant genotype of MV in
Kunming area was H1a, which was consistent with the
genotype of the most common strain in Yunnan province. While the
age range of measles patients was large, the disease mainly
occurred in patients aged <2 years and the majority of patients
had not been vaccinated against measles.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81100091 and
81200067), the Shanghai Municipal Commission of Health and Family
Planning, China (grant nos. 20134Y33 and 2010Y145), Young Medical
Talents Training Program of Pudong Health Bureau of Shanghai, China
(grant no. PWRq2011-02) and the Research Fund of Science and
Technology Commission of Shanghai Municipality (grant no.
14140903600).
References
|
1
|
Jansen VA, Stollenwerk N, Jensen HJ,
Ramsay ME, Edmunds WJ and Rhodes CJ: Measles outbreaks in a
population with declining vaccine uptake. Science. 301:8042003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu Wz, Shui TJ, Li L, et al: Analysis on
epidemiological characteristics and control measures of measles in
China during 2004–2006. Chinese J Vaccines Immun. 5:0002006.
|
|
3
|
Bieback K, Lien E, Klagge IM, Avota E,
Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter
Meulen V and Schneider-Schaulies S: Hemagglutinin protein of
wild-type measles virus activates toll-like receptor 2 signaling. J
Virol. 76:8729–8736. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vidalain PO, Azocar O, Lamouille B, Astier
A, Rabourdin-Combe C and Servet-Delprat C: Measles virus induces
functional TRAIL production by human dendritic cells. J Virol.
74:556–559. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Palosaari H, Parisien JP, Rodriguez JJ,
Ulane CM and Horvath CM: STAT protein interference and suppression
of cytokine signal transduction by measles virus V protein. J
Virol. 77:7635–7644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bellini WJ and Rota PA: Genetic diversity
of wild-type measles viruses: Implications for global measles
elimination programs. Emerg Infect Dis. 4:29–35. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Patterson JB, Thomas D, Lewicki H,
Billeter MA and Oldstone MB: V and C proteins of measles virus
function as virulence factors in vivo. Virology. 267:80–89. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moss WJ and Griffin DE: Global measles
elimination. Nat Rev Microbiol. 4:900–908. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang X, Glendening C, Linke H, Parks CL,
Brooks C, Udem SA and Oglesbee M: Identification and
characterization of a regulatory domain on the carboxyl terminus of
the measles virus nucleocapsid protein. J Virol. 76:8737–8746.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Longhi S, Receveur-Bréchot V, Karlin D,
Johansson K, Darbon H, Bhella D, Yeo R, Finet S and Canard B: The
C-terminal domain of the measles virus nucleoprotein is
intrinsically disordered and folds upon binding to the C-terminal
moiety of the phosphoprotein. J Biol Chem. 278:18638–18648. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
WHO: New genotype of measles virus and
update on global distribution of measles genotypes. Wkly Epidemiol
Rec. 80:347–351. 2005.(In English, French). PubMed/NCBI
|
|
12
|
Zhang Y, Ji YX and Zhu Z: Circulating
pattern analysis for endemic measles viruses in mainland of China.
Zhongguo Yi Miao He Mian Yi. 15:97–103. 2009.(In Chinese).
PubMed/NCBI
|
|
13
|
Xu W, Tamin A, Rota JS, Zhang L, Bellini
WJ and Rota PA: New genetic group of measles virus isolated in the
People's Republic of China. Virus Res. 54:147–156. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Forcic D, Ivancic J, Baricevic M, Mahovlic
V, Tesovic G, Bozinovic D, Margan Gjenero I and Mazuran R: Genetic
characterization of wild type measles virus isolated in Croatia
during the 2003–2004 outbreak. J Med Virol. 75:307–312. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liffick SL, Thoung Thi N, Xu W, Li Y, Lien
Phoung H, Bellini WJ and Rota PA: Genetic characterization of
contemporary wild-type measles viruses from Vietnam and the
People's Republic of China: Identification of two genotypes within
clade H. Virus Res. 77:81–87. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shen XM and Wang W: Pediatrics. 7th.
People's Medical Publishing House; Beijing: pp. 188–191. 2008,
PubMed/NCBI
|
|
17
|
Liu YJ, Li PP, Zhao KX, Wang BJ, Jiang CY,
Drake HL and Liu SJ: Corynebacterium glutamicum contains
3-Deoxy-D-arabino-heptulosonate 7-phosphate synthases that display
novel biochemical features. Appl Environ Microbiol. 74:5497–5503.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moss WJ, Scott S, Ndhlovu Z, Monze M,
Cutts FT, Quinn TC and Griffin DE: Suppression of human
immunodeficiency virus type 1 viral load during acute measles.
Pediatr Infect Dis J. 28:63–65. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Muscat M, Shefer A, Ben Mamou M, Spataru
R, Jankovic D, Deshevoy S, Butler R and Pfeifer D: The state of
measles and rubella in the WHO European Region, 2013. Clin
Microbiol Infect. 20 Suppl 5:S12–S18. 2014. View Article : Google Scholar
|
|
20
|
Update of the nomenclature for describing
the genetic characteristics of wild-type measles viruses: New
genotypes and reference strains. Wkly Epidemiol Rec. 78:229–232.
2003.(In English, French). PubMed/NCBI
|
|
21
|
Mosquera Mdel M, de Ory F, Moreno M and
Echevarría JE: Simultaneous detection of measles virus, rubbela
virus, and parvovirus B19 by using multiplex PCR. J Clin Microbiol.
40:111–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kouomou DW and Wild TF: Adaptation of
wild-type measles virus to tissue culture. J Virol. 76:1505–1509.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cooper LT Jr: Giant cell myocarditis:
Diagnosis and treatment. Herz. 25:291–298. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leonard VH, Sinn PL, Hodge G, Miest T,
Devaux P, Oezguen N, Braun W, McCray PB Jr, McChesney MB and
Cattaneo R: Measles virus blind to its epithelial cell receptor
remains virulent in rhesus monkeys but cannot cross the airway
epithelium and is not shed. J Clin Invest. 118:2448–2458.
2008.PubMed/NCBI
|
|
25
|
Iwasaki M, Takeda M, Shirogane Y, Nakatsu
Y, Nakamura T and Yanagi Y: The matrix protein of measles virus
regulates viral RNA synthesis and assembly by interacting with the
nucleocapsid protein. J Virol. 83:10374–10383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ji Y, Xu W, Zhang Y, Zhu Z, Jiang X, Liang
Y, Zhou S, Zheng J, Chen H, Zhang J, et al: Genetic
characterization of the wild-type measles viruses isolated in six
provinces of China in 2005. Chinese J Virol. 21:407–415. 2005.(In
Chinese).
|
|
27
|
Ding ZR: The Measles Epidemiological
Characteristics in Yunnan Province in 1989–2001. Zhongguo Yi Miao
He Mian Yi. 8:262–263. 2002.(In Chinese).
|