Introduction
Traumatic brain injury (TBI) is usually caused by
external mechanical force (1), such
as falls, motor vehicle accidents, collisions, contact sports and
assaults. Physical, behavioral, emotional and psychosocial changes
may be observed in groups subjected to TBI (2). TBI is known to occur as the result of
two phases: Initial neuronal injury followed by secondary injury.
Initial neuronal injury occurs immediately and is due to the
inciting traumatic event, which is not easily treatable. However,
the second phase occurs from multiple neuropathological processes
and evolves over a period of min to days (3–5). The
delayed nature of secondary injuries allows for medical and
surgical intervention and has become a major focus of TBI treatment
(6). The secondary injuries are
multiple, interacting and interdependent cascades of biological
reactions caused by the initial injury (1). A series of complex biochemical process
associated with the secondary injury occur after a traumatic brain
injury (7,8). In this phase, astrocyte foot process
swelling may result in the damage of the blood-brain barrier
(9). The injuries to the central
nervous system, such as the proliferation of astrocytes, may also
be detected, which may result in a reversal of glutamate uptake and
neuronal depolarization (10,11).
Polyunsaturated fatty acids (PUFAs) are able to
maintain Ca2+ ion and energy homeostasis (12), decrease cognitive deficits and
enhance learning ability during aging (13), and improve the prognosis of ischemic
injury, Alzheimer's disease and Parkinson's disease (14). Docosahexaenoic acid (DHA) is a kind
of polyunsaturated fatty acid, which is predominantly extracted
from deep-sea fishes and algae (15). DHA is not only an important
polyunsaturated fatty acid in the central nervous system, but it is
also the main constituent of n-3 polyunsaturated fatty acids in
cell membranes of cortical gray matter neurons (16). There is limited research on the
impact of supplementation with PUFAs on TBI (17–19).
Nevertheless, these studies (17–19) have
consistently demonstrated the protective effects of PUFAs against
behavioral deficits and cellular degeneration.
DHA has a negative effect on damaging factor
production, such as inflammatory cytokines and free radicals
(20,21). The present study characterized the
impact of DHA supplementation on cognitive function and
inflammatory responses following fluid percussion injury (FPI) in
rats.
Materials and methods
Animal groups
A total of 80 7-week old Sprague-Dawley rats
(male/female ratio 1:1) weighing 300–500 g were maintained in a
temperature (21–25°;C) -and humidity (45–50%)-controlled room with
a 12-h light/dark cycle with ad libitum access to food and
water. Following 1 week of acclimation, the rats were randomly
divided into four groups (male/female ratio 1:1): i) A TBI-model
group (post-TBI with saline; n=20); ii) a sham-operated group
treated with saline (n=20); iii) a TBI-low DHA group, treated with
a low dose of DHA post-TBI (n=20; 370 mg/kg/day DHA); and iv) a
TBI-high DHA group, treated with a high dose of DHA post-TBI (n=20;
740 mg/kg/day DHA). The present study was approved by the Ethics
Committee of Yantai Yuhuangding Hospital (Yantai, China).
Animal models of TBI
Rats were anesthetized by injection of 10% chloral
hydrate (300 mg/kg; Sinopharm Chemical Reagent Co., Ltd., Shanghai,
China), administered intraperitoneally. A 4.5-mm diameter window in
the bone was made 3.5-mm posterior to the bregma and 2.5-mm lateral
to the sagittal suture, without injuring the dura mater. The head
of the rat was positioned in a stereotaxic alignment instrument for
FPI. For FPI, rats were connected through a craniotomy to a
fluid-filled chamber with a small opening; a swinging pendulum hit
one end of the chamber to generate a water pulse that impacted the
exposed brain at the other end of the chamber. Depending on the
location of the craniotomy, the injury could be delivered to the
side of the brain (lateral FPI) or the midline (central FPI), and
the craniotomy was performed at the same location in all of the
rats to the midline. Injury intensity was controlled by adjusting
the height from which the pendulum was dropped (22). Sham-operated rats underwent
craniotomy without FPI (23).
DHA application
Following TBI injury, rats were randomized to three
groups: TBI-model group, TBI-low DHA group, TBI-high DHA group
(n=20 per group). DHA (Sigma-Aldrich; Merck KGaA; Darmstadt,
Germany) was given by intragastric administration. The sham and TBI
model group received equal volumes of saline treatment (0.9% NaCl;
1 ml/kg). Treatment administration began 30 min after TBI injury
and continued once a day for 15 days.
Beam-walking tests
To evaluate complex motor movements and
coordination, beam-walking tests were performed 1 day prior to TBI
and on days 2, 7 and 15 post-TBI (24). The beam was a wooden bar 1,390 mm in
length and 21 mm wide, and was placed 430 mm above the floor. There
was a black box (250×200 mm) at the right end of the beam. A wall
was placed 30 cm to the left of the beam, as rats are more willing
to walk when a wall is placed next to the beam. A mirror was behind
the beam on the side wall. Starting at 2 days before TBI, rats were
habituated to walk on the beam. A rat was put into the box for 1
min. Then, the rat was put onto the beam at a starting distance of
15 cm from the box. The rat was allowed to go to the box and stay
there for 1 min. Thereafter, the rat was put on the beam at a
starting distance of 35 cm from the box. The rat was allowed to go
into the box (and stayed for 1 min). This step was repeated. On the
following day, the rat was put into the box for 1 min and then
allowed to go to the box starting from 35 cm, followed by 70 cm and
finally from a 100-cm distance from the box. On the testing day,
the rat was allowed to cross the whole beam three times. Between
each run, the rat was in the box for 1 min. Scoring was as follows:
0=the rat fell down; 1=the rat was unable to traverse the beam but
remained sitting across the beam; 2=the rat fell down during its
walk; 3=the rat was able to traverse the beam, but the affected
hindlimb did not aid in forward locomotion; 4=the rat traversed the
beam with >3 foot slips; 5=the rat crossed the beam with 1–3
foot slips; and 6=the rat crossed the beam with no foot slips. A
mean score of the three runs for each day was calculated.
Morris water maze trials
The water maze test was initiated at day 15 after
the induction of TBI. The rat was placed on the platform submerged
below the water for 20 sec to allow orientation to extra-maze cues.
The rat was then placed in the water tank at one of four designated
entry points (west, north, east and south) facing the wall and the
time taken to reach the hidden platform was recorded for each
trial. In addition, swimming speed and path length (swimming
distance) were measured. A probe test was conducted 15 days after
TBI, during which the platform was removed. Rats were allowed to
swim for 60 sec to allow evaluation of their memory of the platform
location. The time spent in the four quadrants of the maze was
recorded. A total of eight trials per day were averaged for each
rat and the mean score was calculated for swimming on days 16 and
17 post-TBI. The tracks from all tests were analyzed for a series
of behavioral parameters using SMART 3.0 (Panlab; Barcelona,
Spain).
Histopathological evaluation
Brain specimens were collected and fixed in 10%
formalin for 24 h at room temperature, embedded in paraffin.
Sections of 4-mm thickness were cut from formalin-fixed tissues and
stained with hematoxylin and eosin (10 min for hematoxylin staining
and 5 min eosin staining at room temperature). Specimens were
examined under a light microscope (magnification, ×200).
Western blot analysis
Rats were deeply anesthetized with isoflurane and
perfused transcardially with saline following administration of DHA
for 15 days. The injured brain tissues were collected and
homogenized in radioimmunoprecipitation assay buffer (89900; Thermo
Scientific, Inc., Waltham, MA, USA) containing protease inhibitor
cocktail (P2714) and protease inhibitor mixture (P2714; both
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Homogenates were
centrifuged at 13,000 × g at 4°C for 30 min. The supernatant was
saved to determine its protein concentration by Bradford assay.
Total protein (50 µg/lane) was separated by 10% SDS-PAGE and then
blotted onto a polyvinylidene difluoride membrane. Following
blocking (2 h at room temperature) with ProteinFree T20 Blocking
Buffer (37573; lot no. LB141635; Thermo Scientific, Inc.),
membranes were incubated for 1 h at room temperature with the
following primary antibodies: Rabbit monoclonal anti-cleaved
caspase-3 antibody (1:1,000; 9664; Cell Signaling Technology, Inc.,
Danvers, MA, USA), rabbit anti-B-cell lymphoma 2 (Bcl-2; 1:1,000;
2872; Abcam, Cambridge, MA, USA), rabbit anti-Bcl-2-associated X
protein (Bax; 1:1,000; 2772; Abcam) and rat monoclonal anti-β-actin
polyclonal antibody (1:2,000; A2228; Sigma-Aldrich; Merck KGaA).
The membranes were subsequently incubated with horseradish
peroxidase-conjugated secondary antibody at room temperature for 40
min (1:5,000; goat anti-rabbit, ZB-2301; goat anti-mouse, ZDR5307;
ZSGB-BIO Technology Co., Ltd., Beijing, China). Protein bands were
visualized using an enhanced chemiluminescence reagent (EMD
Millipore; Billerica, MA, USA) and quantified by densitometry using
a Genomic and Proteomic Gel Documentation System from Syngene
(Frederick, MD, USA). The protein band intensities of Bcl-2, Bax
and caspase-3 were normalized by the corresponding band intensities
of β-actin from the same samples to control for loading errors. The
results from animals under various experimental conditions were
then normalized by mean values of the corresponding control
animals.
Statistical analysis
Statistical analysis was performed using SPSS 19.0
software (IBM Corp., Armonk, NY, USA) for Windows. All data were
presented as the mean ± standard deviation. The significance of
differences between groups was evaluated using one-way analysis of
variance followed by Dunnett's test. P<0.05 was considered to
indicate a statistically significant difference.
Results
DHA protects against motor deficits
induced by TBI
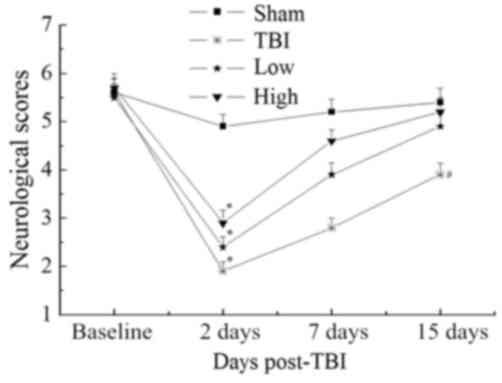
Beam-walking trials were performed to evaluate
complex motor movements and coordination. The results are
summarized in Fig. 1. Further
analysis at each time point did not reveal any significant
differences in beam-walking between the groups at baseline
(P>0.05). All groups were significantly impaired at 2 days
post-TBI compared with the sham group (P<0.05). Both the TBI-low
and TBI-high DHA groups improved over the 15-day follow-up. The
changes observed over time in the TBI-model group were less
notable. At 15 days post-TBI, the performance of the TBI-high and
-low DHA groups approached that of the sham group (P>0.05);
however, the neurological scores of the TBI model and TBI-low DHA
groups were significantly lower than those of the sham group
(P<0.05).
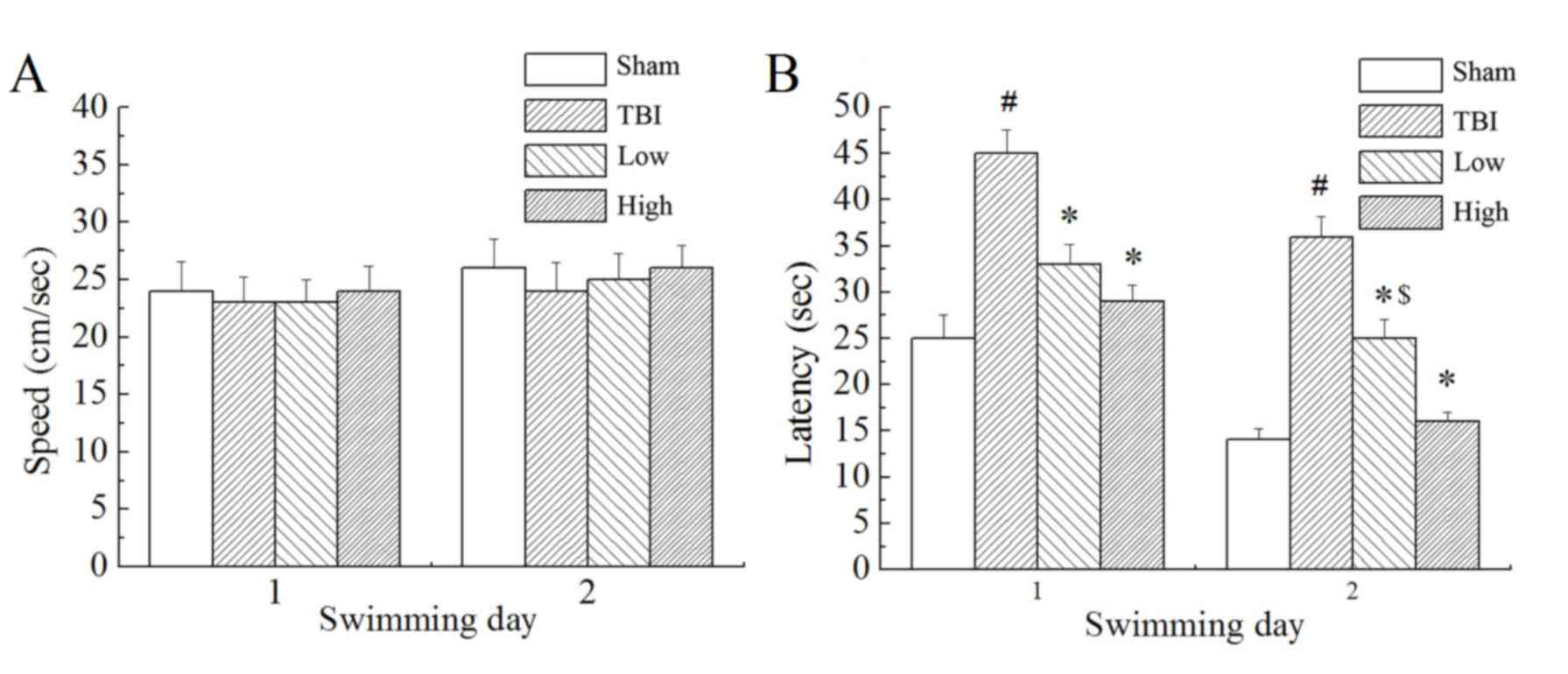
Morris water maze trials
In the Morris water maze testing, the rats were made
to find the target platform to escape from swimming in the pool
with water. Results demonstrated that there was no significant
difference in swimming speed between the groups (Fig. 2A). However, there were significant
differences in escape latency (time taken to find the submerged
platform; P<0.05; Fig. 2B) and
marked differences in swimming distance (not shown) between the
groups. The TBI model group demonstrated significantly longer
escape latencies and swimming distances than the sham group
(P<0.05). Compared with the TBI model group, the TBI-low and
TBI-high DHA groups demonstrated significantly shorter latency
times (P<0.05). Compared with the TBI-low DHA group, the
TBI-high DHA group demonstrated shorter escape latencies and
swimming distances.
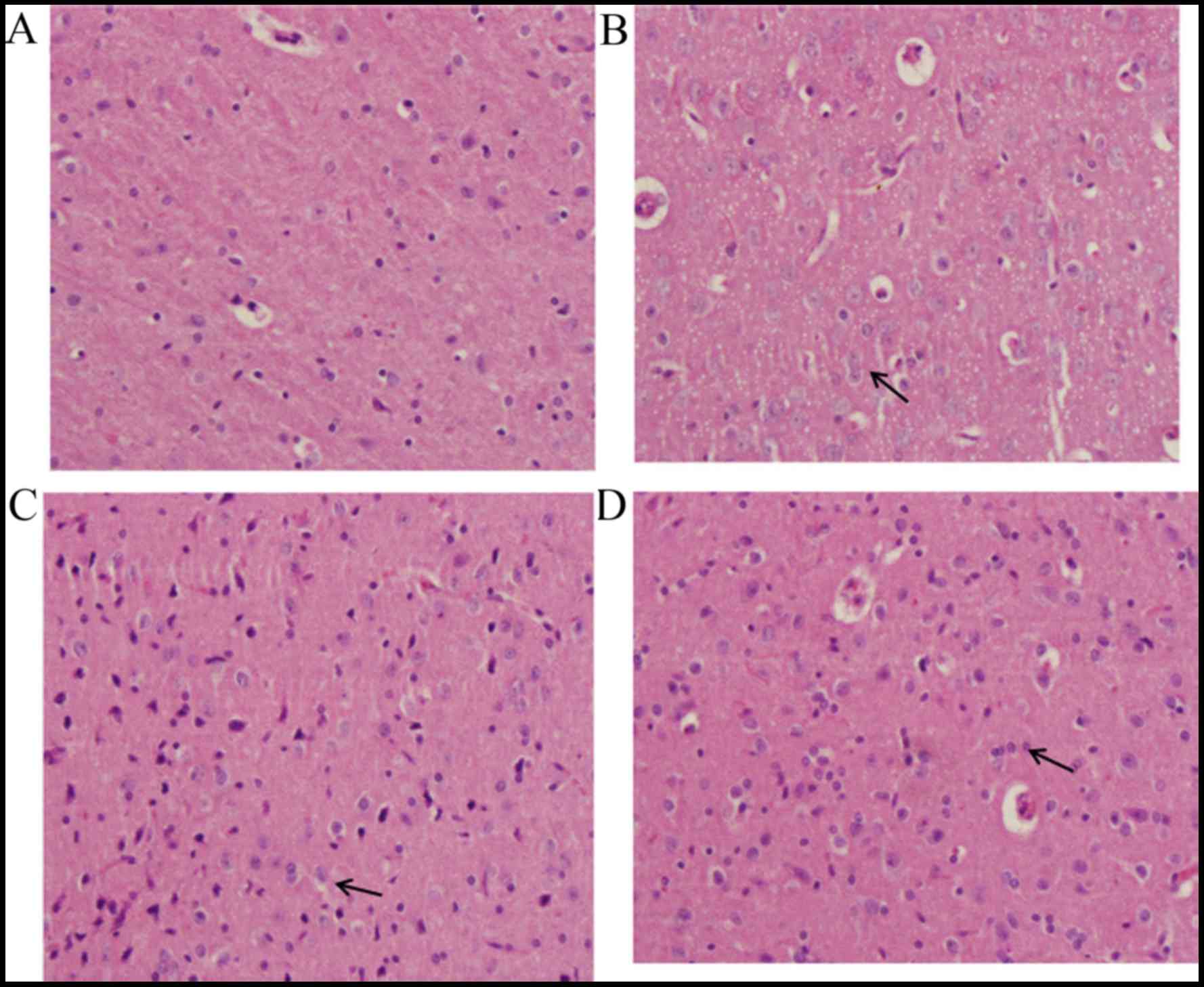
Pathological findings of brain
tissue
In the sham group, the majority of cells in the
brain tissue demonstrated normal morphology (Fig. 3A). Evidently damaged nerve cell
structure, swelling of nerve cells, shrinking of nucleolus, obvious
hyperemia and congestion in blood capillaries, the proliferation of
glial cells and the formation of clusters of neurons and the
‘satellite phenomenon’ in glial cells were observed in the
TBI-model group (Fig. 3B). The
changes observed in the DHA-treated groups were milder than those
of the TBI-model group, which appeared to be dose-dependent. The
group treated with the high dose of DHA demonstrated basically
normal structure of brain tissue, slight hyperemia and congestion
in blood capillaries and the proliferation of glial cells (Fig. 3C). While the group treated with the
low dose of DHA (Fig. 3D)
demonstrated obvious pathological changes compared with the high
DHA-treated group.
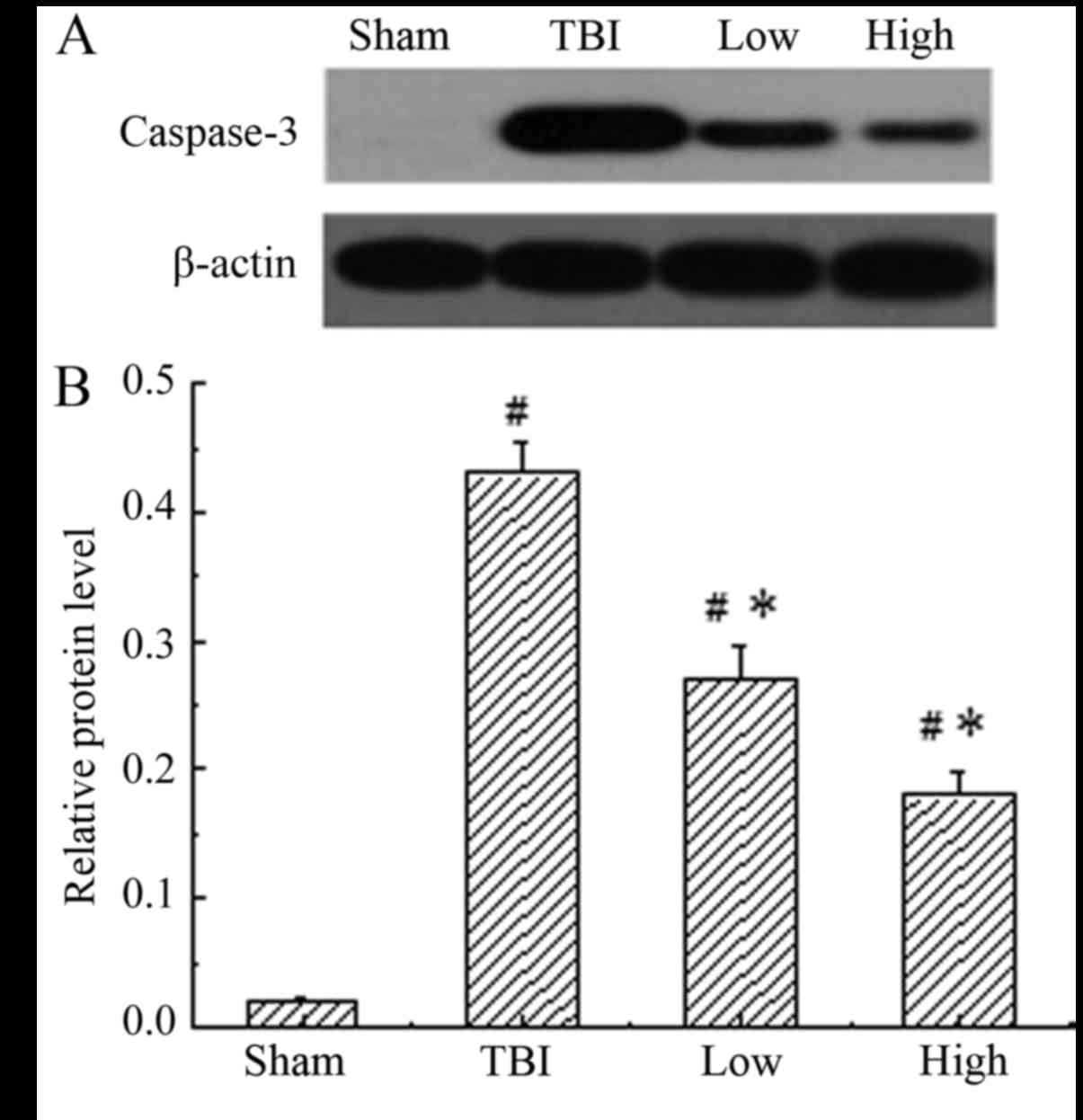
DHA inhibits the expression of
caspase-3
Caspase-3 was constitutively expressed in the
sham-operated group, while the TBI-model group (post-TBI with
saline) demonstrated a significant increase in the expression of
caspase-3 compared with the sham group (P<0.05). When treated
with DHA, the expression of caspase-3 was significantly reduced
compared with that in the rats of the TBI model groups (P<0.05),
indicating that DHA inhibited the expression of caspase-3 (Fig. 4). The inhibitory effect of DHA was
more obvious at a high dosage.
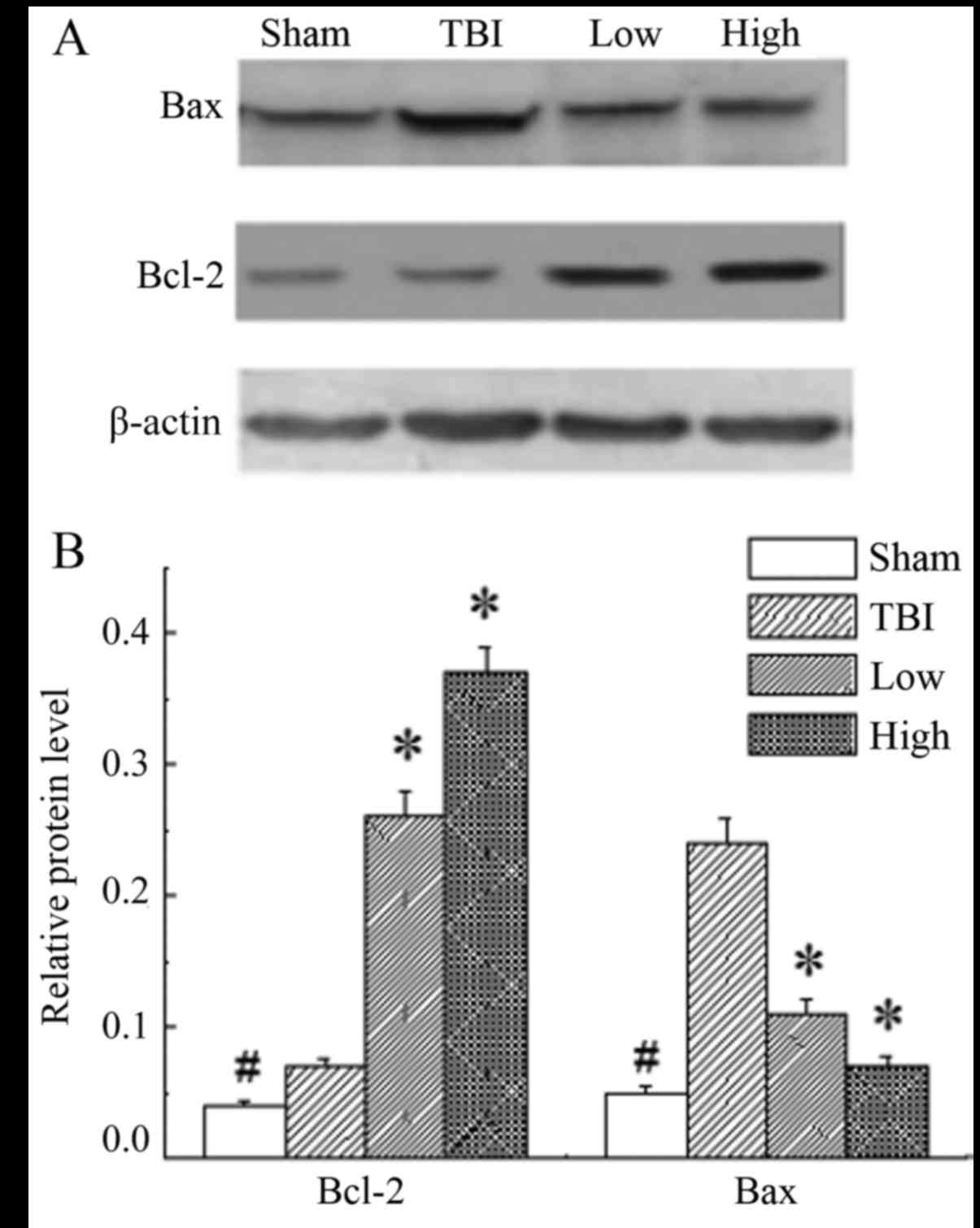
DHA rescues neurons by upregulating
the Bcl-2:Bax ratio
In the rats of the sham group, the expression level
of Bcl-2 and Bax was significantly lower than those in the other
groups (P<0.05). The results demonstrated significant
upregulation of Bcl-2 and downregulation of Bax in the DHA groups
compared with the TBI-model group (P<0.05; Fig. 5).
Discussion
Traumatic brain injury is a prevalent neurological
disorder that results in gray and white matter injury (25). In the present study, 15 days of
supplementation with DHA elicited robust protection against
sensorimotor and cognitive deficits in a rat model of TBI. This
protective dietary strategy has previously demonstrated much
success in other paradigms (26,27). In
the beam-walking test, further analysis at each time point did not
reveal any significant differences in beam-walking between the
groups at baseline, which suggested that learning ability was
maintained in the injured group. All groups were impaired
significantly on day 2 post-TBI compared with the sham group,
followed by a gradual increase thereafter. However, a significant
increase in score was observed in rats that received DHA relative
to the TBI-model rats, suggesting that the administration of DHA
significantly improved the motor movements and coordination of
rats. Analogously, the rats that received treatment with DHA
performed better than TBI-model rats in Morris water maze trials.
All TBI-related groups demonstrated longer escape latencies and
swimming distances than the sham group. Compared with the TBI-low
DHA group, the TBI-high DHA group had shorter escape latency and
swimming distances. According to a study by Bailes and Mills
(28), supplementation with DHA
significantly decreased amyloid precursor protein-positive axons in
the white matter tract.
In models of TBI, pro-apoptotic mechanisms may be
activated during secondary damage to promote caspase-3-mediated
cell death (29). In the present
study, it was demonstrated that the expression of active caspase-3
was downregulated in the cortex of rats treated with DHA following
TBI, which protected the cortical neurons from apoptosis. Caspase-3
activation in cell death signaling may occur via three routes: The
mitochondrial route (related to Bcl-2 and Bax), the endoplasmic
reticulum route or a death receptor route involving FAS and FAS
ligand (30). Bcl-2 gene families
have been identified to be regulators of apoptosis. Of these genes,
Bcl-2 is an anti-apoptotic protein that serves as a critical
regulator of pathways involved in apoptosis. Contrastingly, Bax is
a pro-apoptotic protein, which controls the integrity of the
mitochondrial outer membrane (31).
Research has indicated that the Bcl-2 protein physically interacts
with several of its homologous proteins, forming heterotypic dimers
(32). The Bcl-2/Bax dimerization is
considered critical for interactions during apoptosis (33). Bcl-2 is downregulated in the injured
brain (34) and the overexpression
of Bcl-2 reduces post-ischemic injury (35). Chronic daily administration of DHA
significantly increased Bcl-2 expression in brain tissues in the
present study.
To determine the signal that modulated the
activation of caspase-3 following TBI, the present study measured
the protein expression levels of Bcl-2 and Bax in order to
calculate the Bcl-2:Bax ratio. The results demonstrated the
upregulation of Bcl-2 and the downregulation of Bax protein levels
following treatment with DHA. Therefore, these results indicated
that DHA may increase the Bcl-2:Bax ratio to inhibit the expression
of caspase-3, ultimately protecting neurons from apoptosis.
In conclusion, the present study demonstrated that
DHA supplementation was a viable strategy to mitigate injury caused
by TBI. DHA treatment improved memory function, which may be due to
its anti-inflammatory properties. Furthermore, DHA was useful in
preventing neuronal damage following brain ischemia. The present
data support the belief that fish oil supplementation in humans may
exert similar prophylactic or preventive actions against neuronal
damage in the future.
References
|
1
|
Maas AI, Stocchetti N and Bullock R:
Moderate and severe traumatic brain injury in adults. Lancet
Neurol. 7:728–741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lauterbach MD, Notarangelo PL, Nichols SJ,
Lane KS and Koliatsos VE: Diagnostic and treatment challenges in
traumatic brain injury patients with severe neuropsychiatric
symptoms: Insights into psychiatric practice. Neuropsychiatr Dis
Treat. 11:1601–1607. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McIntosh TK: Neurochemical sequelae of
traumatic brain injury: Therapeutic implication. Cerebrovasc Brain
Metabol Rev. 6:109–162. 1994.
|
|
4
|
Park E, Bell JD and Baker AJ: Traumatic
brain injury: Can the consequences be stopped. CMAJ. 178:1163–1170.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raghupathi R, Conti AC, Graham DI,
Krajewski S, Reed JC, Grady MS, Trojanowski JQ and McIntosh TK:
Mild traumatic brain injury induces apoptotic cell death in the
cortex that is preceded by decreased in cellular Bcl-2
immunoreactivity. Neuroscience. 110:605–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McHugh GS, Engel DC, Butcher I, Steyerberg
EW, Lu J, Mushkudiani N, Hernández AV, Marmarou A, Maas AI and
Murray GD: Prognostic value of secondary insults in traumatic brain
injury: Results from the IMPACT study. J Neurotrauma. 24:287–293.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
King JT Jr, Carlier PM and Marion DW:
Early glasgow outcome scale scores predict long-term functional
outcome in patients with severe traumatic brain injury. J
Neurotrauma. 22:947–954. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fork M, Bartels C, Ebert AD, Grubich C,
Synowitz H and Wallesch CW: Neuropsychological sequelae of diffuse
traumatic brain injury. Brain Inj. 19:101–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bao HJ, Wang T, Zhang MY, Liu R, Dai DK,
Wang YQ, Wang L, Zhang L, Gao YZ, Qin ZH, et al: Poloxamer-188
attenuates TBI-induced blood-brain barrier damage leading to
decreased brain edema and reduced cellular death. Neurochem Res.
37:2856–2867. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lenzlinger PM, Morganti-Kossmann MC,
Laurer HL and McIntosh TK: The duality of the inflammatory response
to traumatic brain injury. Mol Neurobiol. 24:169–181. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liou AK, Clark RS, Henshall DC, Yin XM and
Chen J: To die or not to die for neurons in ischemia, traumatic
brain injury and epilepsy: A review on the stress-activated
signaling pathways and apoptotic pathways. Prog Neurobiol.
69:103–142. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu A, Ying Z and Gomez-Pinilla F: The
salutary effects of DHA dietary supplementation on cognition,
neuroplasticity and membrane homeostasis after brain trauma. J
Neurotrauma. 28:2113–2122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cole GM, Ma QL and Frautschy SA: Dietary
fatty acids and the aging brain. Nutr Rev. 68:S102–111. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bousquet M, Calon F and Cicchetti F:
Impact of ω-3 fatty acids in Parkinson's disease. Ageing Res Rev.
10:453–463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kalmijn S, Feskens EJ, Launer LJ and
Kromhout D: Polyunsaturated fatty acids, antioxidants and cognitive
function in very old men. Am J Epidemiol. 145:33–41. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dyall SC: Long-chain omega-3 fatty acids
and the brain: A review of the independent and shared effects of
EPA, DPA and DHA. Front Aging Neurosci. 7:522015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu A, Ying Z and Gomez-Pinilla F: Omega-3
fatty acids supplementation restores mechanisms that maintain brain
homeostasis in traumatic brain injury. J Neurotrauma. 24:1587–1595.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang PK, Khatchadourian A, McKinney RA
and Maysinger D: Docosahexaenoic acid (DHA): A modulator of
microglia activity and dendritic spine morphology. J
Neuroinflammation. 12:342015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pusceddu MM, Kelly P, Stanton C, Cryan JF
and Dinan TG: N-3 polyunsaturated fatty acids through the lifespan:
Implication for psychopathology. Int J Neuropsychopharmacol.
19:pyw0782016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen W, Esselman WJ, Jump DB and Busik JV:
Anti-inflam-matory effect of docosahexaenoic acid on
cytokine-induced adhesion molecule expression in human retinal
vascular endothelial cells. Invest Ophthalmol Vis Sci.
46:4342–4347. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Florent S, Malaplate-Armand C, Youssef I,
Kriem B, Koziel V, Escanyé MC, Fifre A, Sponne I, Leininger-Muller
B, Olivier JL, et al: Docosahexaenoic acid prevents neuronal
apoptosis induced by soluble amyloid-beta oligomers. J Neurochem.
96:385–395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gyoneva S and Ransohoff RM: Inflammatory
reaction after traumatic brain injury: Therapeutic potential of
targeting cell-cell communication by chemokines. Trends Pharmacol
Sci. 36:471–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McIntosh TK, Vink R, Noble L, Yamakami I,
Fernvak S, Soares H and Faden AL: Traumatic brain injury in the
rat: Characterization of a lateral fluid-percussion model.
Neuroscience. 28:233–244. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohlsson AL and Johansson BB: Environment
influences functional outcome of cerebral infarction in rats.
Stroke. 26:644–649. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gasparovic C, Yeo R, Mannell M, Ling J,
Elgie R, Phillips J, Doezema D and Mayer AR: Neurometabolite
concentrations in gray and white matter in mild traumatic brain
injury: An 1H-magnetic resonance spectroscopy study. J Neurotrauma.
26:1635–1643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguemeni C, Delplanque B, Rovère C,
Simon-Rousseau N, Gandin C, Agnani G, Nahon JL, Heurteaux C and
Blondeau N: Dietary supplementation of alpha-linolenic acid in an
enriched rapeseed oil diet protects from stroke. Pharmacol Res.
61:226–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martín A, Boisgard R, Kassiou M, Dollé F
and Tavitian B: Reduced PBR/TSPO expression after minocycline
treatment in a rat model of focal cerebral ischemia: A PET study
using (18)F]DPA-714. Mol Imaging Biol. 13:10–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bailes JE and Mills JD: Docosahexaenoic
acid reduces traumatic axonal injury in a rodent head injury model.
J Neurotrauma. 27:1617–1624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clark RS, Kochanek PM, Watkins SC, Chen M,
Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL
and Graham SH: Caspase-3 mediated neuronal death after traumatic
brain injury in rats. J Neurochem. 74:740–753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salakou S, Kardamakis D, Tsamandas AC,
Zolota V, Apostolakis E, Tzelepi V, Papathanasopoulos P, Bonikos
DS, Papapetropoulos T, Petsas T and Dougenis D: Increased Bax/Bcl-2
ratio up-regulates caspase-3 and increases apoptosis in the thymus
of patients with myasthenia gravis. In Vivo. 21:123–132.
2007.PubMed/NCBI
|
|
31
|
Solaroglu I, Tsubokawa T, Cahill J and
Zhang JH: Anti-apoptotic effect of granulocyte-colony stimulating
factor after focal cerebral ischemia in the rat. Neuroscience.
143:965–974. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hara A, Hirose Y, Wang A, Yoshimi N,
Tanaka T and Mori H: Localization of Bax and Bcl-2 proteins,
regulators of programmed cell death, in the human central nervous
system. Virchows Arch. 429:249–253. 1996.PubMed/NCBI
|
|
33
|
Xiao D and Zhang L: Upregulation of Bax
and Bcl-2 following prenatal cocaine exposure induces apoptosis in
fetal rat brain. Int J Med Sci. 5:295–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kao TK, Ou YC, Kuo JS, Chen WY, Liao SL,
Wu CW, Chen CJ, Ling NN, Zhang YH and Peng WH: Neuroprotection by
tetramethylpyrazine against ischemic brain injury in rats.
Neurochem Int. 48:166–176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei L, Cui L, Snider BJ, Rivkin M, Yu SS,
Lee CS, Adams LD, Gottlieb DI, Johnson EM Jr, Yu SP and Choi DW:
Transplantation of embryonic stem cells overexpressing Bcl-2
promotes functional recovery after transient cerebral ischemia.
Neurobiol Dis. 19:183–193. 2005. View Article : Google Scholar : PubMed/NCBI
|