Introduction
Hepatic encephalopathy (HE), presenting as
functional dysregulation in the central nervous system, is a
cognitive syndrome induced by liver diseases and characterized by
dysbiosis in the metabolism (1). The
1-year survival rate of HE patients has been reported to be 42%,
while the 3-year survival rate declines to 23% (1). Approximately 30% of cirrhotic patients
develop minimal HE (MHE), which displays no apparent symptoms, but
results in a higher risk of developing overt HE and of mortality
(2). Therefore, diagnosis and
treatment at the MHE stage is of crucial significance in reducing
the mortality rates in liver cirrhosis patients. A comprehensive
understanding of MHE pathogenesis and associated biomarkers in its
diagnosis and treatment is, thus, of increasing importance.
Several hypotheses have been proposed for the
pathogenesis of HE, including toxicosis by ammonium, manganese,
false neurotransmitters and imbalance in plasma insulin and amino
acids (1–6). The patients in the present study were
split into alcoholic and non-alcoholic groups because liver
cirrhosis may be induced by alcoholism, while certain liver
cirrhotic patients develop MHE. MHE can also be caused by various
pathological conditions, including an accumulation of mercaptans
and short-chain fatty acids (7).
Numerous of these hypotheses are associated with dysbiosis in the
ammonium metabolism; therefore, ammonium toxicosis is regarded as
one of the most important inducing factor of HE (4). Certain urease-producing gut microbes,
including Klebsiella, Proteus and Helicobacter
pylori, have been demonstrated to be associated with blood
ammonium levels and are implicated in the pathogenesis of HE.
Therefore, targeting these microbes for therapeutic purposes is of
great potential for the treatment of HE. For instance, probiotics
and prebiotics are used to lower the gut pH and prevent the growth
of the urease-producing bacteria (8). In addition, MHE has been reported to be
a risk factor for motor vehicle accidents due to attention deficit
caused by MHE patients (9), while a
recent study suggested that yoghurt consumption was an independent
negative risk factor for traffic accidents in patients with liver
cirrhosis (10). Furthermore,
antibiotics are also typically employed to control the
proliferation of Helicobacter pylori (11). However, no reports currently exist on
the effect of combined treatment with antibiotics (such as
rifaximin, used in the present study) and probiotics on gut
microbiota alterations. Besides, alcoholic and non-alcoholic MHE
patients may also respond differently to treatment in terms of gut
microbiota alterations. Therefore, investigation into these aspects
will reveal how gut microbiota are differentially regulated by
different pathogenic causes and therapeutic regimens, and therefore
provide guidance for the drugs used to target the specific
pathogenic causes.
As an increasing number of patients with chronic
hepatitis is reported in the southwestern Yunnan in China,
particularly patients infected with hepatitis C virus, the
incidence of liver cirrhosis with or without HE is likely to
increase. Given the correlation between the metabolic functions of
gut microbiota and HE, the aim of various therapeutic regimens is
to restore the gut microbiota towards the normal composition and
functions.
Therefore, the present study conducted the first
comprehensive metagenomics investigation into gut microbiota
alterations subsequent to treatment with rifaximin, a
well-documented anti-MHE drug, or a combination treatment
consisting of rifaximin and probiotics (12). The comparison of the two treatments
investigated in the current study provides a deeper insight into
the effect of different treatments on gut microbiota. Furthermore,
the study subjects were grouped into alcoholic and non-alcoholic
MHE patients, reporting the different responses observed in these
patients and therefore providing guidance for the design of more
effective treatment regimens.
Patients and methods
Patients
A total of 14 MHE patients from the Affiliated
Hospital of Kunming University of Science and Technology, (Kunming,
China) were recruited into the present investigation. The
characteristics of these patients are presented in Table I. Liver cirrhotic patients, induced
by alcoholism, hepatitis B virus infection or other causes, were
recruited initially. These patients were then diagnosed using
multiple testing, including number connection test (NCT) and digit
symbol test (DST). In the NCT, 25 numbers were distributed on a
piece of paper, and patients were required to connect the same
numbers within the shortest time possible. Mistakes were corrected
in a timely manner and the test continued. The total time required
to complete the assessment was recorded. In the DST, a matrix of
nine symbols were provided to symbolize the numbers 1–9, and
patients were required to fill the corresponding symbols underneath
the numbers in the shortest time possible. Scores were calculated
according to the number of correct answers and adjusted according
to the age range. Scores of ≤7 were considered to be abnormal.
Patients with abnormal scores in both NCT and DST were diagnosed as
MHE patients and recruited as study subjects. Patients that were
diagnosed as alcoholic liver cirrhosis were grouped into the
alcoholic category, whereas liver cirrhotic patients that were
induced by other factors, including hepatitis B virus infection,
were grouped into the non-alcoholic category.
 | Table I.Patient characteristics and
treatment. |
Table I.
Patient characteristics and
treatment.
| Patient | Sex | Age, years | Cause of
cirrhosis | Year of
diagnosis | DST, min | Treatment |
|---|
| 1 | Male | 65 | Autoimmune
hepatitis | 2011 | 46 | RP |
| 2 | Male | 57 | Hepatitis B
cirrhosis | 2011 | 34 | R |
| 3 | Female | 48 | Primary biliary
cirrhosis | 2012 | 10 | R |
| 4 | Female | 55 | Autoimmune
hepatitis | 2011 | 13 | RP |
| 5 | Female | 46 | Hepatitis B
cirrhosis | 2010 | 35 | RP |
| 6 | Male | 48 | Hepatitis
B/alcoholic | 2011 | 31 | RP |
| 7 | Male | 41 | Autoimmune
hepatitis | 2006 | 50 | RP |
| 8 | Female | 43 | Autoimmune
hepatitis | 2008 | 50 | R |
| 9 | Male | 39 | Hepatitis B
cirrhosis | 2009 | 21 | R |
| 10a | Male | 57 | Alcoholic
cirrhosis | 2012 | 26 | RP |
| 11a | Male | 47 | Alcoholic
cirrhosis | 2011 | 26 | R |
| 12a | Male | 60 | Alcoholic
cirrhosis | 2011 | 48 | RP |
| 13a | Male | 68 | Alcoholic
cirrhosis | 2007 | 35 | R |
| 14a | Male | 63 | Alcoholic
cirrhosis | 2009 | 43 | R |
All procedures were conducted with informed consent
from the patients and in consistency with ethical requirement of
the ethics board of Affiliated Hospital of Kunming University of
Science and Technology.
Treatment
Patients were treated with rifaximin or with
rifaximin (Nanjing Pharmaceuticals Co., Ltd., Nanjing, China) and
probiotics on a random basis. Rifaximin tablets (400 mg) were
administered orally twice a day, and samples were collected 4 weeks
after the treatment. The probiotic (trade name, Meichangan) formula
included live combined Bacillus subtilis and Enterococcus
faecium enteric-coated 250-mg capsules (Hanmi Pharmaceutical
Co., Ltd., Beijing, China) containing 0.5 billion live bacteria
each.
Sample collection
Samples were collected under sterile conditions
using sputum collection box. Faeces were flash frozen in liquid
nitrogen 2 h after collection and preserved at −80°C. DNA from the
intestinal bacteria community of patients was isolated immediately
prior to treatment and 4 weeks following treatment with rifaximin
or rifaximin plus probiotics, using the CTAB
(hexadecyltrimethylammonium bromide) approach (13). Agarose gel electrophoresis (0.8%) was
performed to assess DNA concentration and purity, followed by
dilution to 1 ng/µl. Next, DNA samples were subjected to
metagenomic analysis in order to obtain information on the changes
in the composition of bacterial species following treatment.
Sequencing strategy of bacterial
samples
In order to determine the bacterial composition in
the subjects' intestines, polymerase chain reaction was employed to
amplify the highly variable V3, V4 and V5 regions of bacterial 16S
rRNA using specifically designed primers (F515,
5′-GTGCCAGCMGCCGCGGTAA-3′ and R806, 5′-GGACTACVSGGGTATCTAAT-3′; M
stands for A/C, V stands for A/C/G, and S stands for C/G) as
previously described (14). While in
conventional testing only the V4 region of 16S rRNA is amplified,
the present study investigated more regions, therefore greatly
enhancing the sensitivity and specificity of the approach.
DNA was extracted from samples, followed by quality
validation, as previously described (14). Qualified DNA was used as a template
for the amplification of the V3-V5 region of 16S rRNA. Following
purification, amplicons were subjected to analysis with MiSeq
system according to the manufacturer's instructions (Illumina,
Inc., San Diego, CA, USA) to obtain primary sequences. Adapters and
low-quality reads were removed from the primary sequences, followed
by multiple bioinformatics analysis, including operational
taxonomic unit (OTU), abundance, α and β diversity, and clustering
analyses.
OTU analysis and statistics at various
taxonomic levels
Original sequences were filtered, as previously
described (14) to remove
low-quality reads. Valid reads were subjected to the UCLUST-based
clustering methodology in QIIME software (version 2; Scikit-Bio
open source; http://qiime.org/). Sequences with
>95% similarity were clustered as one OTU, denoting one
bacterial species. The taxonomic levels included order, family,
genus and species. All procedures were completed as previously
described (14).
α diversity analysis
α diversity is used to describe the abundance of
various species in the sample. Based on OTU analysis, rarefaction
curves were generated using R to demonstrate the observed number of
OTUs and the estimated number of species (Chao1 index) with the
increase of read numbers (15).
Chao1 index is used to estimate the number of OTUs or species in
the population, therefore symbolizing the diversity of the
population. Shannon diversity indices of various samples were also
calculated to assess the diversity and distribution evenness of the
species. This index differs from others in that it takes into
account the distribution evenness of the species. Therefore, these
indices can denote the diversity and distribution of the species in
a given population. The detailed analytical method was performed as
previously described (15).
β diversity analysis
β diversity, represented by the principal coordinate
analysis (PcoA), reveals the magnitude of community composition and
describes the alterations in species distribution in various major
coordinates. PcoA using weighted Unifrac from the 28 samples
calculated the values in three principal components- PC1, PC2 and
PC3. PcoA analysis then locates the samples in plots against three
principal coordinates to showcase the relative similarities and
abundances of the samples. The principal coordinates stand for a
matrix of major components, which account for 39.67, 22.38 and
11.99% of the microbiota composition, respectively. The dot plot
provided a direct image of differences in the intestinal microbiota
of each subject post-treatment compared with pre-treatment by
examining at the distance between these samples. The detailed
analytical method was performed as previously described (15).
Clustering analysis
Based on the data of distribution at the phylum and
genus level, clustering was conducted using the matrix of
unweighted and weighted Unifrac, as previously described (15). Fewer branches present between the two
samples indicate that they are closer in species distribution.
Results
Study patients
A total of 14 MHE patients were recruited in the
present investigation. Among these, 7 patients were treated with
rifaximin alone, while the remaining 7 were treated with rifaximin
in conjugation with probiotics. When grouping according to the
alcohol consumption of patients, 9 patients were defined as
non-alcoholic MHE patients, whereas the remaining 5 were
categorized as alcoholic MHE patients. Table I gives the basic information of the
patients and their treatment. Patients prior to treatment were
denoted as numbers 1–14 and after treatment, the patients were
denoted as the number+treatment. For instance, 1+RP stands for
sample from patient 1 after Rifaximin plus Probiotics treatment,
whereas 2+R stands for sample from patient 2 after Rifaximin
treatment.
Basic data set collection and
analysis
In total, 28 samples from the 14 study subjects
prior to and following treatment yielded a data set consisting of
1,585,825 high-quality classifiable 16S rRNA gene sequences, with a
mean of 56,637 sequences per sample. Using the conventional
criterion of 95% sequence similarity (corresponding to
taxonomically valid species), a total of 4,456 OTUs were
identified, with a mean of 159 OTUs per sample. These OTUs were
divided into 10 genera. The top four genera included
Lactobacillus and Bacteroides, which were the two
dominant genera, as well as Streptococcus and
Clostridium.
Clinical treatment generally reduced
gut microbiota diversity in patients
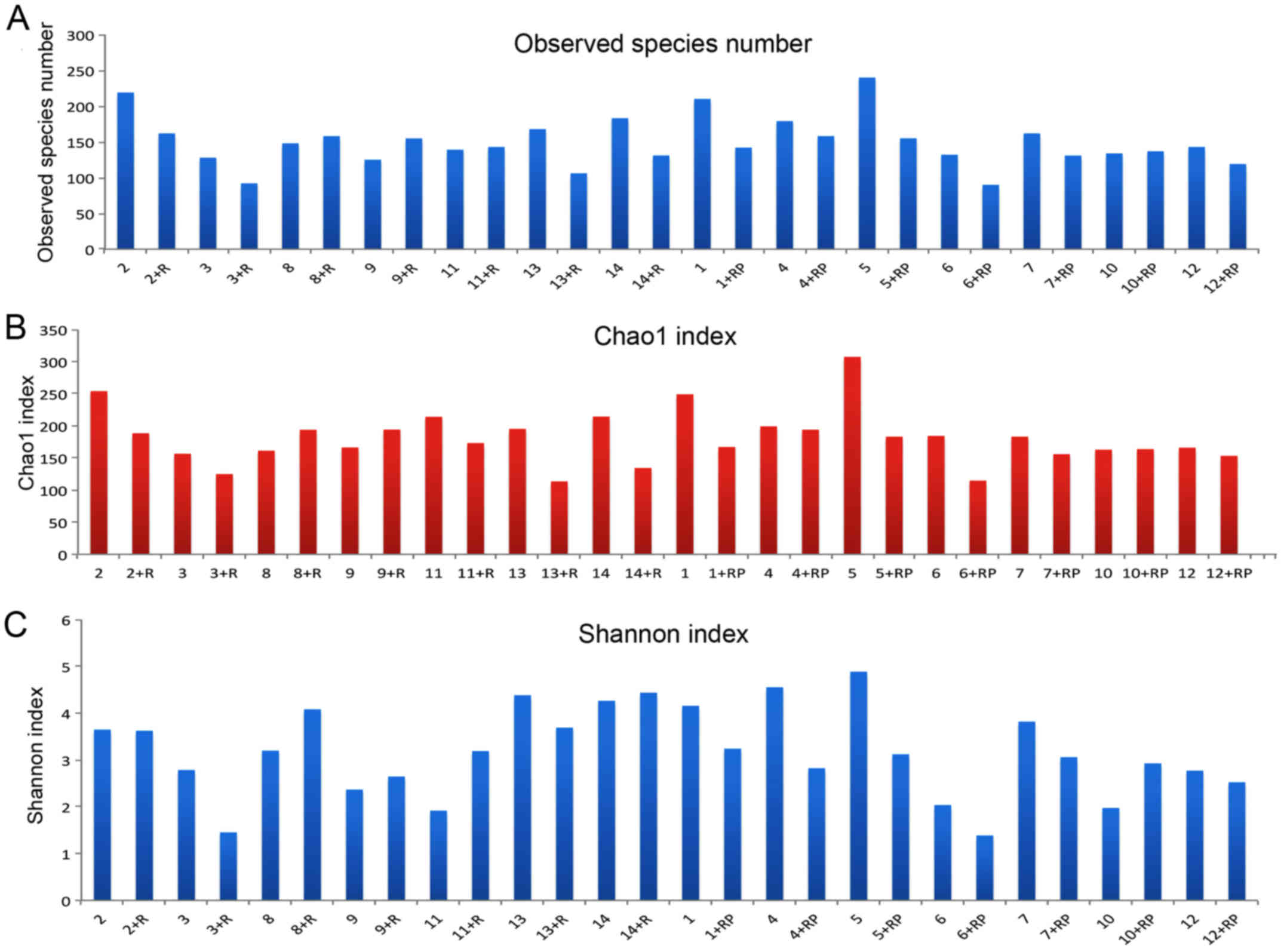
The number of observed species in each sample with
the increment in sequence numbers is shown in Fig. 1A. An overall reduction in the actual
numbers of observed species was observed post-treatment. For
instance, the species number in patient 5 was decreased following
treatment with rifaximin. In contrast, certain patients presented
the opposite trend, such as patient 8 who had slightly higher
species number post rifaximin treatment. The marginal difference in
estimated species numbers prior to and following treatment with
rifaximin plus probiotics appeared to be smaller compared with that
in patients treated with rifaximin alone.
The Chao1 index was calculated to estimate the total
number of OTUs based on the actual observed species number. An
overall decrease in Chao1 index was detected subsequent to
treatment, although certain exceptions were observed (Fig. 1B). Changes in Chao1 index before and
after treatment and the differences in Chao1 index between the two
treatments are presented in Fig. 1B
and roughly corresponded to the trend detected for species numbers
(Fig. 1A).
Shannon index is shown in Fig. 1C. By comparing the Shannon index of
each patient prior to and following treatment, a general decline in
the index was observed following treatment, with a few exceptions,
such as patient 10. Upon taking a closer look, a predominant
reduction in the index can be observed following treatment with
rifaximin plus probiotics, whereas the group with rifaximin only
treatment presented a more diversified response. Furthermore,
certain patients, such as patient 3, presented a reduced Shannon
index following rifaximin treatment, while others presented the
opposite effect, such as patient 8. In addition, patient 2 did not
have an evident response to rifaximin treatment in terms of
microbiota diversity, as Shannon index remained almost the same
subsequent to treatment.
Similarly, if subjects are divided into the
alcoholic and non-alcoholic MHE groups, a predominant decrease in
Shannon index is observed in non-alcoholic patients following
treatment. By contrast, alcoholic liver cirrhosis patients
demonstrated divergent responses, with patient 11 presenting a
higher index, and patients 12 and 13 exhibiting lower values post
treatment.
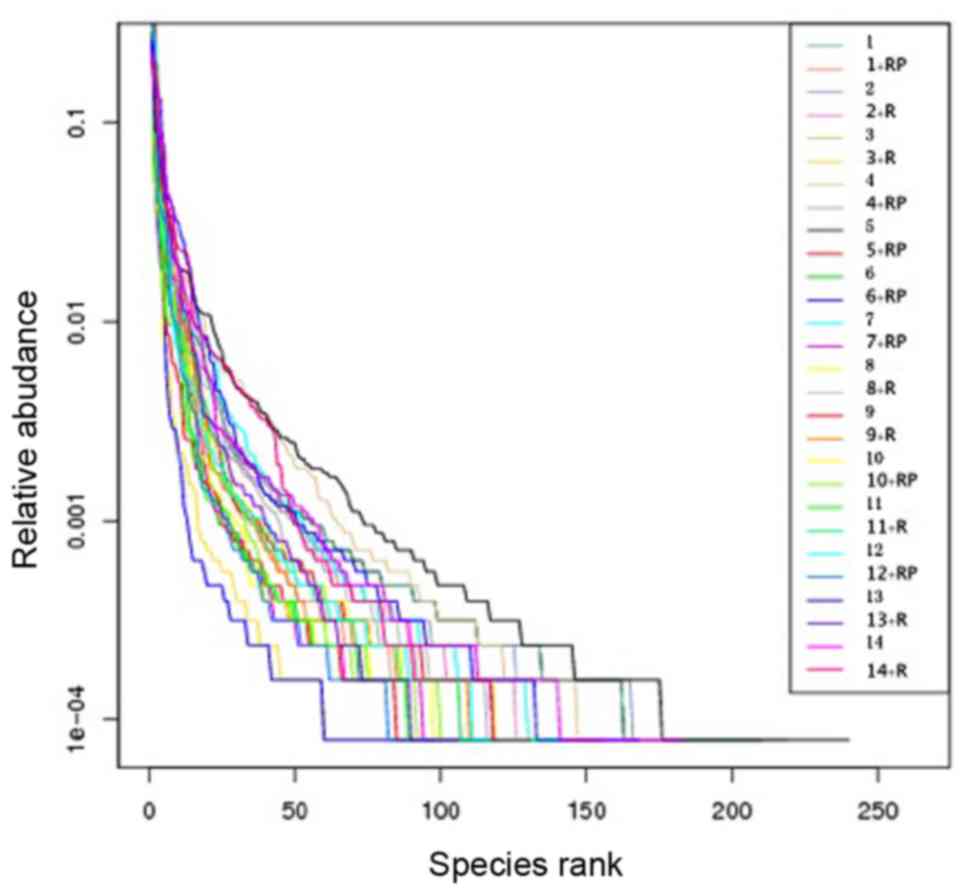
Rank abundance of the samples also
revealed reduced diversity following treatment
Rank abundance, presented in Fig. 2, visualizes the species richness and
evenness in the sample. The total number of species is demonstrated
by the maximum reading of each curve on the x-axis. In general,
these maximal × values in Fig. 2 are
higher in samples with higher observed species numbers (Fig. 1A) and higher Chao1 index (Fig. 1B). Species evenness, as deduced from
the slope of the curves, was generally higher in samples with a
higher number and more homogeneous distribution of species.
Alcohol addiction compromises
treatment efficacy
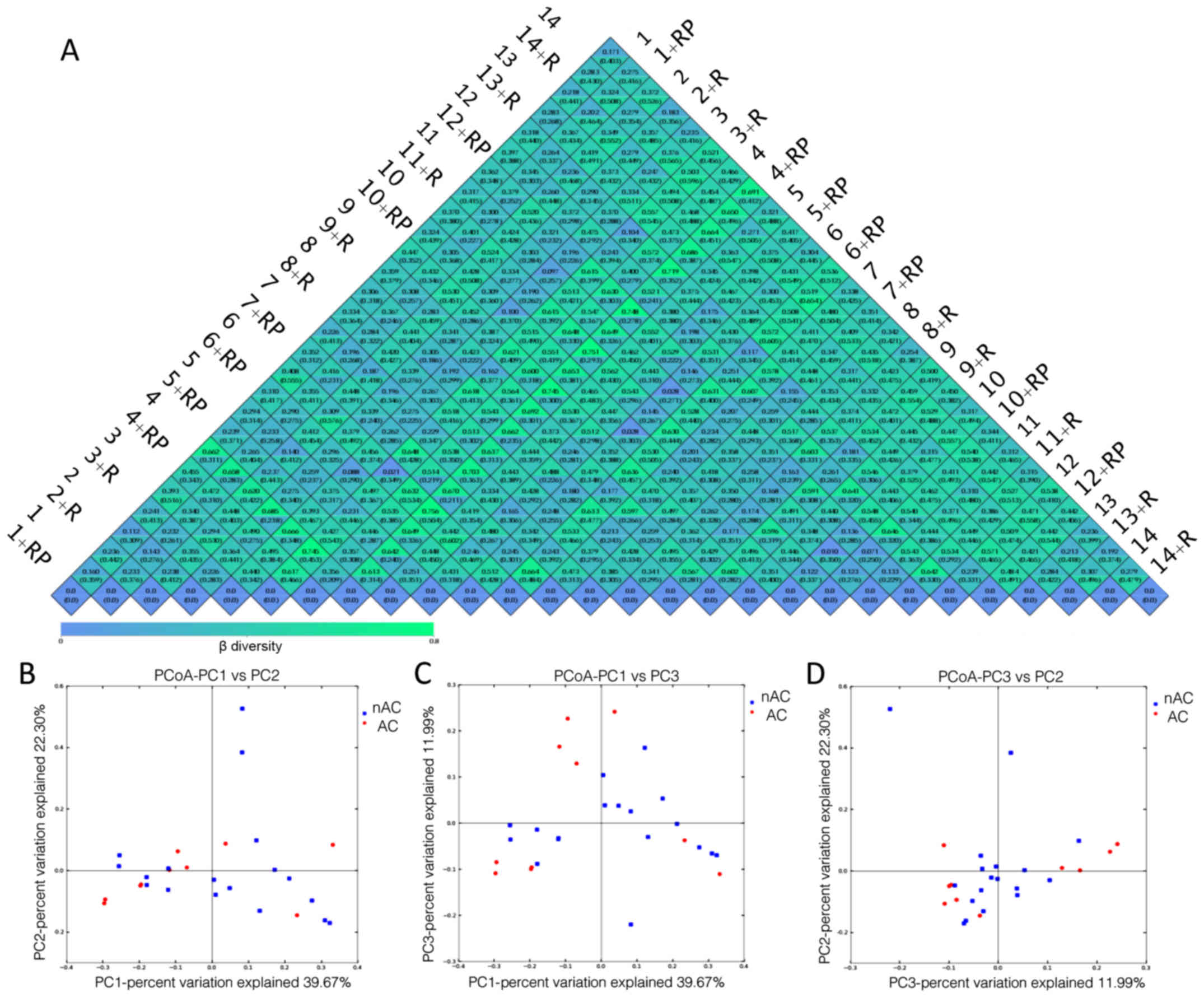
Fig. 3A presents a
heatmap of the results of β diversity analysis with all the
weighted (top value in each box) and unweighted (bottom value in
each box) Unifrac values between two patients according to pairwise
comparison, prior to or following treatment. Comparing the values
for each patient prior to and following treatment, the magnitude of
response to the treatment for that specific patient is obtained.
For instance, the weighted Unifrac for patient 11 pre- and
post-rifaximin treatment was 0.133, demonstrating the least
response to the treatment. By contrast, a large difference in
microbiota was observed in patient 9, with a weighted UniFrac value
of 0.602. Furthermore, no notable difference was observed between
the two groups receiving different treatment. However,
non-alcoholic MHE patients displayed generally higher Unifrac
values as compared with alcoholic MHE patients.
Weighted PcoA analysis calculates the values in
three principal components-PC1, PC2 and PC3. It then locates the
samples in plots against the three principal coordinates to
showcase the relative similarities and abundances of the samples.
The principal coordinates stand for a matrix of major components,
which account for 39.67, 22.38 and 11.99% of the microbiota
composition, respectively (Fig.
3B-D). It was also observed that non-alcoholic MHE patients
presented better clustering in these principal coordinates (PC1,
PC2 and PC3; with only a few distant exceptions), in contrast to
the more scattered pattern observed for alcoholic patients.
Treatment leads to altered abundance
in certain major phyla and genera
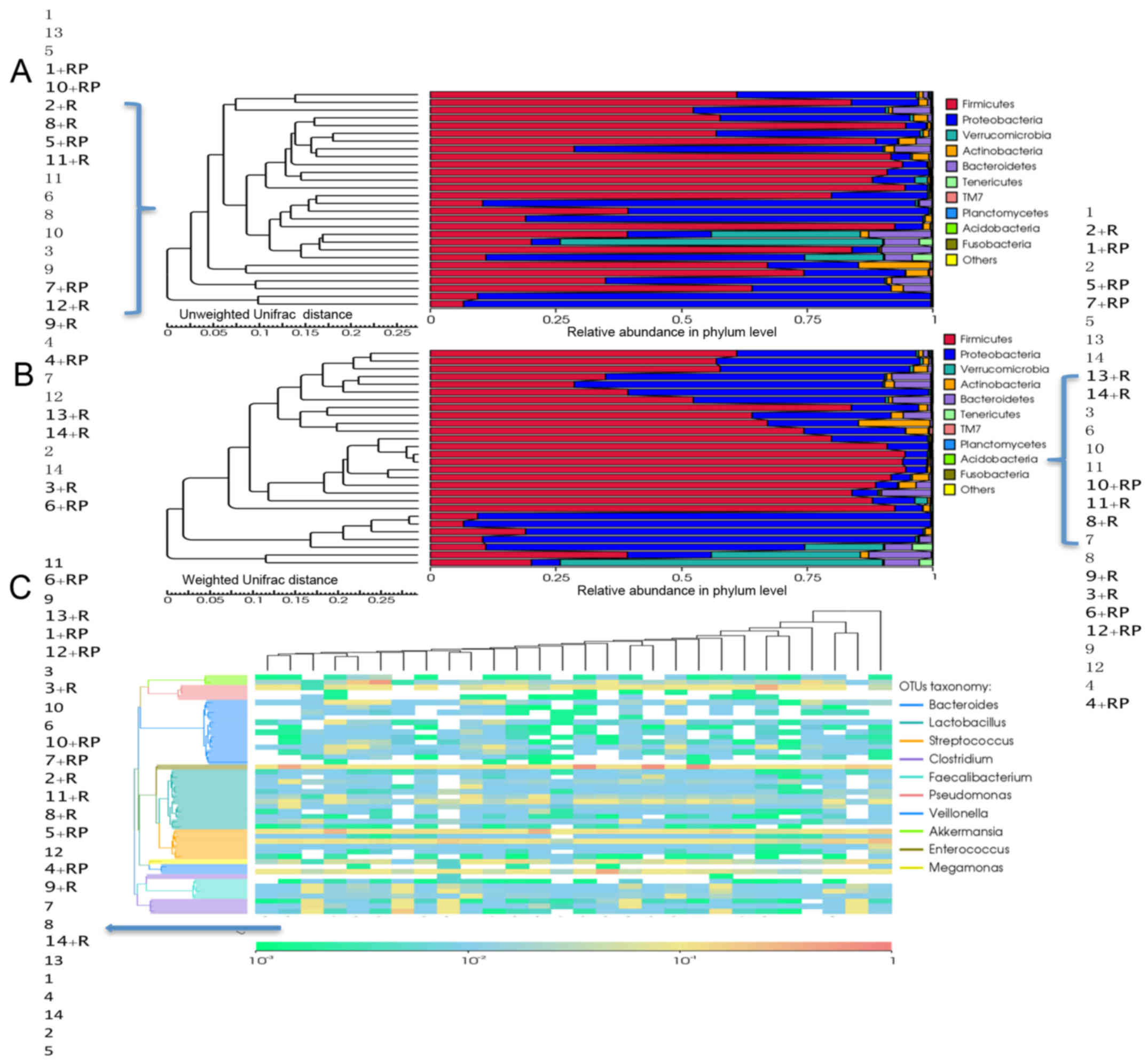
Bar charts in Fig. 4A and
B display the relative abundance of gut microbiota at the
phylum level, while samples are clustered according to unweighted
and weighted Unifrac. The results demonstrated that Firmicutes and
Proteobacteria constitute the majority of the gut microbiota. In
general, a decrease in the abundance of Firmicutes was observed in
the patients following treatment. The trend was more apparent in
non-alcoholic patients, with alcoholic patients demonstrating
unaltered or even increased numbers, such as patients 12 and 14. In
contrast, Proteobacteria, which constituted the second largest
phylum in the gut microbiota, exhibited a divergent trend, with its
abundance increasing post-treatment in 7 out of the 14 patients.
The remaining half of the patients demonstrated unaltered or
decreased abundance of Proteobacteria. No correlation was detected
between the two different treatment groups.
A clustering tree is a method that clusters the
samples based on the unweighted and weighted Unifracs. In the
clustering tree, the closer two samples are located, the more
similar their microbiota compositions are. By examining the
clustering trees using unweighted and weighted Unifracs to showcase
the microbiota similarities between the patients prior to and after
the treatment, a much shorter distance was observed in each patient
prior to and following treatment in the weighted tree as
represented by the number of connection lines between bars in
Fig. 4A and B. This indicates
reduced pairwise disparity pre- and post-treatment due to the
relative abundances of bacterial species considered as weights.
When these weights (relative species abundances) are taken into
account, the calculated weighted Unifracs are closer to each other,
leading to shortened distances. For instance, patient 13 presented
a greater unweighted UniFrac distance in comparison with the
weighted distance.
Clustering analysis at the genus level for each
sample was also performed (Fig. 4C).
By comparing the abundance prior to and following treatment for
each patient, a predominant reduction in Clostridium
abundance was observed post-treatment, with a concurrent increase
in Lactobacillus and decrease in Streptococcus and
Faecalibacterium abundances in a small fraction of the
patients. Particularly, reduction in one Streptococcus
species was detected in only 2 patients post-treatment, which
belonged to the non-alcoholic and combinatorial treatment with
rifaximin plus probiotics groups. Bacteroides demonstrated a
divergent trend, with certain patients presenting higher abundance
of Bacteroides post-treatment, such as patient 1, and
certain others having decreased abundance, such as patient 2.
However, no correlation was observed between the change patterns
and grouping criteria.
Discussion
The present study provided an insight into the
varying response of alcoholic and non-alcoholic MHE patients to
different treatments, including rifaximin alone or rifaximin plus
probiotics, in terms of the gut microbiota composition. The current
results demonstrated an overall decline in gut microbiota diversity
following treatment, which was more apparent in MHE patients
treated with rifaximin and probiotics. In addition, non-alcoholic
MHE patients responded better, presenting a decreased microbiota
diversity and ammonia-producing bacteria abundance, compared with
alcoholic patients.
Gut microbiota is critical in maintaining normal
intestinal functions, including digestion, absorption, nutrition
supply and immune activation (16–19).
Distinctive gut microbiota alterations are connected with the
cognitive and inflammatory status in HE patients with liver
cirrhosis (20,21). A thorough understanding of their
roles in the pathogenesis of HE, particularly in MHE, is critical
in the identification of appropriate strategies targeting the
microbial species in order to restore the normal microbiota
composition and functions. Given that gut microbiota is diverse in
different populations, studies targeting a specific population are
required in order to more precisely decipher the pathogenesis and
identify therapeutic strategies. The southwestern Yunnan Province
in China hosts a large population of hepatitis B and C patients,
giving rise to increased number of MHE patients. Therefore,
investigation into gut microbiota alterations in these patients may
reveal distinct mechanisms of the disease pathogenesis and
facilitate treatment strategy development specifically for this
population.
Certain treatment regimens have been designed for
the therapy of MHE, including administration of rifaximin (a
semisynthetic antibiotic), probiotics, lactulose, prebiotics and
synbiotics (22). The study by Bajaj
et al (22) demonstrated no
significant microbiota alteration subsequent to rifaximin
treatment, with the exception of a modest decrease in
Veillonellaceae and increase in Eubacteriaceae. However, rifaximin
administration contributed to cognitive functions by shifting the
networks centered on Enterobacteriaceae, Porphyromonadaceae and
Bacteroidaceae from pathogenic to beneficial metabolite linkages.
Despite all these advances, there is a lack of mechanistic
investigations on combination treatments in terms of their impact
on gut microbiota. Therefore, the present study aimed to reveal the
gut microbiota alterations in MHE patients treated with rifaximin
plus probiotics, as compared with rifaximin alone. Further insight
into how alcoholic and non-alcoholic MHE patients may respond to
these two regimens was also examined.
In the current study, a general decline in gut
microbiota diversity was observed when the MHE patients were
treated with rifaximin alone or rifaximin plus probiotics. The
difference in microbiota composition was also signified by the
paired UniFrac of each MHE patient prior and subsequent to
treatment in β diversity analysis. This can be explained by the
nature of rifaximin, which is an antibiotic intended to kill
certain microbes, such as E. coli, thereby reducing
diversity. MHE patients treated with rifaximin plus probiotics
yielded an overall lower magnitude of decrease in the estimated
species number following the treatment as compared with the
pre-treatment value. Probiotics, which are microorganisms
considered to be beneficial for a more balanced microorganism
distribution in the gut when consumed, may account for this
disparity (23). However, these
patients demonstrated a more significant reduction in Shannon index
following treatment. As Shannon index considers the relative
abundance of bacterial species, this phenomenon suggests that the
combined treatment of rifaximin plus probiotics may more
significantly distort the relatively balanced distribution of
microbial species abundance rather than reduce the number of
bacterial species. Furthermore, the presence of probiotics improves
the symptoms by shifting the microbiota composition from
pathological to beneficial distribution, thereby creating a more
favorable gut environment to restore the beneficial species and
microbiota functions that may be partially compromised by
rifaximin. However, β analysis did not reveal a considerable
difference in the unweighted or weighted Unifrac value of each MHE
patient between the two treatment groups. The Unifrac values were
calculated based on the phylogenetic tree, or the relative position
of each bacterial species in the evolutionary tree. This suggests
that, despite the impact of probiotics on microbiota diversity,
this impact may be negligible when considering the entire
phylogenetic tree.
Upon comparison of alcoholic and non-alcoholic MHE
patients in the present study, non-alcoholic subjects presented a
predominant reduction in Shannon diversity index and higher
pairwise Unifrac values post treatment vs. pre-treatment values,
when compared to alcoholic MHE patients. Non-alcoholic patients
also presented a more consistent trend in the abundance of certain
major bacterial phyla post-treatment, such as decline in
Firmicutes. Given the much lower abundance of Firmicutes in healthy
individuals in Yunnan (24), it was
suggested that alcoholic MHE patients possess lower capability in
the restoration of gut microbiota and have a reduced response to
the treatment regimen. Similarly, alcoholic MHE patients presented
more scattered weighted PcoA results, suggesting that their
response to treatment may be more unpredictable. Furthermore,
certain non-alcoholic MHE patients demonstrated a decrease
post-treatment in Streptococcus, a urease-producing bacteria
genus, while alcoholic patients did not exhibit this change. The
decrease in Streptococcus may consequently lower ammonia
levels and improve patient conditions.
In the present study, clustering analysis by
weighted and unweighted Unifrac distances produced two distinct
clusters, with shorter weighted Unifrac distances observed in the
majority of the MHE patients pre- and post-treatment. As weighted
Unifrac values take into consideration the relative abundance of
each phylum, these shorter distances suggest a relatively small
change in the abundances of major phyla. By contrast, as unweighted
Unifrac values consider all existing phyla regardless of their
abundance, the longer distances of each patient pre- and
post-treatment in the clusters indicate a marked change in the
composition or the number of microorganisms at the phylum level.
Nevertheless, this does not exclude the differences in the
abundances of major phyla, however subtle they are. Certain
patients even demonstrated considerable alterations in the
composition of these bacteria, such as patient ZFY3. In general,
the abundance of Firmicutes declined following treatment,
particularly in non-alcoholic MHE patients.
Ammonia produced by gut microbes is regarded as an
important inducing agent of MHE, and its level is highly correlated
with MHE pathogenesis (25).
Specific bacterial species carry urease-encoding genes and have
been found to be associated with ammonia metabolism, including
Clostridium, Klebsiella, Proteus,
Veillonella and Helicobacter (8,11). Zhang
et al (26) identified that
Streptococcaceae and Veillonellaceae are enriched in liver
cirrhotic patients with or without MHE, and MHE-unique interplay
pattern of gut microbiota is greatly influenced by these two
bacterial families. Bajaj et al (22) also noted no significant gut
microbiota alteration following rifaximin treatment, with an
exception of a modest decrease in Veillonellaceae and an increase
in Eubacteriaceae. However, the present study did not demonstrate
any evident alterations in the two genera of Streptococcus
and Veillonella post-treatment, with the exception of two
non-alcoholic MHE patients treated with rifaximin plus probiotics,
who presented decreased Streptococcus levels. The
discrepancy may lie in the different taxonomic levels at which
statistical analysis was performed, since the present study
conducted analysis at the genus level, whereas the aforementioned
analysis (22) was performed at the
family level. Another explanation may be that the Veillonellaceae
family is enriched in MHE, but its abundance is not significantly
altered by rifaximin treatment.
The current investigation also detected a robust
decline in the genus of Clostridium, which belongs to the
Firmicutes phylum. Specific Clostridium species are
considered to be hyper-ammonia producing, such as Clostridium
aminophilum and Clostridium histolyticum. The proteases
secreted by Clostridium histolyticum can digest native and
denatured proteins into amino acids with the production of ammonia
(27). Although Clostridium
was not identified to be highly enriched in MHE patients in the
present study its reduction post-treatment may lead to declined
ammonia levels in the blood and thereby reduced severe ammonia
toxicosis, thus contributing to improved cognitive conditions in
the MHE patients. Besides, Clostridium, Streptococcus
and Veillonella, which belong to the Firmicutes phylum and
are ammonia-producing bacteria, contributed to the decline in
Firmicutes at the phylum level, as observed in the current study.
The decline in these bacterial genera post-treatment leads to
partial restoration of the microbiota composition as compared with
healthy individuals, and therefore improvement in clinical
conditions (4).
Lactobacillus, which also belongs to the
Firmicutes phylum, demonstrated an increase in certain patients
following treatment in the present study. This may also beneficial
for the treatment of MHE, since its metabolic product (lactic acid)
decreases gut pH and thereby kills the bacterial species that have
urease to convert nutrition into ammonia. Lactobacillus not
only reduces ammonia levels in the gut, but also creates a
favorable environment for the growth of probiotics, such as
Lactobacillus and Bifidobacterium. Jointly, these
changes contribute to the modulation of gut microbiota dysbiosis
associated with MHE (8).
In conclusion, the present study investigated the
effect of different treatment strategies, including rifaximin alone
or rifaximin plus probiotics, on gut microbiota in MHE patients.
The addition of probiotics in the treatment regimen distorted the
distribution of bacteria in the gut and reduced
Streptococcus abundance. In addition, non-alcoholic MHE
patients presented a higher magnitude of gut microbiota alterations
subsequent to treatment, particularly reduction in the abundance of
Firmicutes.
Acknowledgements
The current study was financially supported by
grants from the National Natural Science Foundation of China (grant
no. 81260077). The authors would like to thank Professor Xiangyang
Kong (Medical College of Kunming University of Science and
Technology, Kunming, China), Professor Zhigang Zhang (Kunming
Institute of Zoology, Chinese Academy of Sciences, Kunming, China)
and Dr Junhong Su (Medical College of Kunming University of Science
and Technology, Kunming, China) for their exceptional technical
assistance and help with the manuscript.
References
|
1
|
Dbouk N and McGuire BM: Hepatic
encephalopathy: A review of its pathophysiology and treatment. Curr
Treat Options Gastroenterol. 9:464–474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kato A, Tanaka H, Kawaguchi T, Kanazawa H,
Iwasa M, Sakaida I, Moriwaki H, Murawaki Y, Suzuki K and Okita K:
Nutritional management contributes to improvement in minimal
hepatic encephalopathy and quality of life in patients with liver
cirrhosis: A preliminary, prospective, open-label study. Hepatol
Res. 43:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amodio P, Montagnese S, Gatta A and Morgan
MY: Characteristics of minimal hepatic encephalopathy. Metab Brain
Dis. 19:253–267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernardini P and Fischer JE: Amino acid
imbalance and hepatic encephalopathy. Annu Rev Nutr. 2:419–454.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Norman K and Pirlich M: Gastrointestinal
tract in liver disease: Which organ is sick? Curr Opin Clin Nutr
Metab Care. 11:613–619. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prakash R and Mullen KD: Mechanisms,
diagnosis and management of hepatic encephalopathy. Nat Rev
Gastroenterol Hepatol. 7:515–525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawaguchi T, Taniguchi E and Sata M:
Effects of oral branched-chain amino acids on hepatic
encephalopathy and outcome in patients with liver cirrhosis. Nutr
Clin Pract. 28:580–588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Riordan SM and Williams R: Gut flora and
hepatic encephalopathy in patients with cirrhosis. N Engl J Med.
362:1140–1142. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawaguchi T, Taniguchi E and Sata M: Motor
vehicle accidents: How should cirrhotic patients be managed? World
J Gastroenterol. 18:2597–2599. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawaguchi T, Suetsugu T, Ogata S, Imanaga
M, Ishii K, Esaki N, Sugimoto M, Otsuyama J, Nagamatsu A, Taniguchi
E, et al: An association between dietary habits and traffic
accidents in patients with chronic liver disease: A data-mining
analysis. Biomed Rep. 4:615–622. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bass NM, Mullen KD, Sanyal A, Poordad F,
Neff G, Leevy CB, Sigal S, Sheikh MY, Beavers K, Frederick T, et
al: Rifaximin treatment in hepatic encephalopathy. N Engl J Med.
362:1071–1081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhan T and Stremmel W: The diagnosis and
treatment of minimal hepatic encephalopathy. Dtsch Arztebl Int.
109:180–187. 2012.PubMed/NCBI
|
|
13
|
Kumar MS, Kaur G and Sandhu AK: Genomic
DNA isolation from fungi, algae, plant, bacteria and human blood
using CTAB. Int J Sci Res. 3:617–618. 2014.
|
|
14
|
Qin J, Li R, Raes J, Arumugam M, Burgdorf
KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al: A
human gut microbial gene catalogue established by metagenomic
sequencing. Nature. 464:59–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kindt R and Coe R: Tree diversity
analysis: A manual and software for common statistical methods for
ecological and biodiversity studies. World Agroforestry Centre.
2005.
|
|
16
|
Khoruts A and Sadowsky MJ: Therapeutic
transplantation of the distal gut microbiota. Mucosal Immunol.
4:4–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wikoff WR, Anfora AT, Liu J, Schultz PG,
Lesley SA, Peters EC and Siuzdak G: Metabolomics analysis reveals
large effects of gut microflora on mammalian blood metabolites.
Proc Natl Acad Sci USA. 106:3698–3703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
MacDonald TT, Monteleone I, Fantini MC and
Monteleone G: Regulation of homeostasis and inflammation in the
intestine. Gastroenterology. 140:1768–1775. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cho I and Blaser MJ: The human microbiome:
At the interface of health and disease. Nat Rev Genet. 13:260–270.
2012.PubMed/NCBI
|
|
20
|
Bajaj JS, Hylemon PB, Ridlon JM, Heuman
DM, Daita K, White MB, Monteith P, Noble NA, Sikaroodi M and
Gillevet PM: Colonic mucosal microbiome differs from stool
microbiome in cirrhosis and hepatic encephalopathy and is linked to
cognition and inflammation. Am J Physiol Gastrointest Liver
Physiol. 303:G675–G685. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bajaj JS, Ridlon JM, Hylemon PB, Thacker
LR, Heuman DM, Smith S, Sikaroodi M and Gillevet PM: Linkage of gut
microbiome with cognition in hepatic encephalopathy. Am J Physiol
Gastrointest Liver Physiol. 302:G168–G175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bajaj JS, Heuman DM, Sanyal AJ, Hylemon
PB, Sterling RK, Stravitz RT, Fuchs M, Ridlon JM, Daita K, Monteith
P, et al: Modulation of the metabiome by rifaximin in patients with
cirrhosis and minimal hepatic encephalopathy. PLoS One.
8:e600422013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rijkers GT, de Vos WM, Brummer RJ, Morelli
L, Corthier G and Marteau P: Health benefits and health claims of
probiotics: Bridging science and marketing. Br J Nutr.
106:1291–1296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuang YS, Li SH, Guo Y, Lu JH, He JR, Luo
BJ, Jiang FJ, Shen H, Papasian CJ, Pang H, et al: Composition of
gut microbiota in infants in China and global comparison. Sci Rep.
6:366662016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jain L, Sharma BC, Srivastava S, Puri SK,
Sharma P and Sarin S: Serum endotoxin, inflammatory mediators, and
magnetic resonance spectroscopy before and after treatment in
patients with minimal hepatic encephalopathy. J Gastroenterol
Hepatol. 28:1187–1193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Z, Zhai H, Geng J, Yu R, Ren H, Fan
H and Shi P: Large-scale survey of gut microbiota associated with
MHE Via 16S rRNA-based pyrosequencing. Am J Gastroenterol.
108:1601–1611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oakley CL and Warrack GH: The alpha, beta
and gamma antigens of Clostridium histolyticum (Weinberg
& Séguin, 1916). J Gen Microbiol. 4:365–373. 1950. View Article : Google Scholar : PubMed/NCBI
|