Introduction
Pancreatic cancer, one of leading causes of
cancer-associated mortality in Western countries, has an extremely
poor prognosis with an overall five-year survival rate of <5%
and a median survival of <1 year (1,2). The
poor prognosis of pancreatic cancer is mainly due to the malignant
behavior of pancreatic cancer, including metastasis, recurrence and
chemoresistance. As major hallmarks of pancreatic cancer, extensive
local invasion, early systemic dissemination and resistance to most
cytotoxic drugs also attribute to its malignancy.
At present, the clinical standard of care for
early-diagnosed or advanced pancreatic cancer is chemotherapy with
2′,2′-difluorodeoxycytidine (dFdC; gemcitabine), a cytotoxic
nucleoside analogue. Gemcitabine has a relatively low tumor
response rate of ~15% and offers a median survival time of 5 months
(3), although it only extends
survival by a mere 5 weeks on average (4). Of note, pancreatic tumors in a
substantial number of patients are already (or rapidly become)
chemoresistant to gemcitabine and display a loss of fundamental
response (2). Thus, improving
chemosensitivity is a strategy for increasing therapeutic effects
on pancreatic cancer. For this purpose, recent studies have
identified several chemoresistance mechanisms associated with the
metabolism and molecular targets of gemcitabine (5,6).
The discovery of cancer stem-like cells (CSCs) has
provided novel insight into carcinogenesis and the effects of
cancer therapy. It has been suggested that sub-populations of CSCs
within solid tumors sustain the formation and growth of the tumor.
The presence of CSCs also accounts for tumor recurrence due to
their self-renewal capacity and metastatic potential (7,8).
Compared with other tumor cells, CSCs also present with a
significantly increased chemoresistance to conventional
therapeutics, including gemcitabine (9). Studies assessing specific oncogene
models of cancer and specific signaling pathways revealed that CSCs
tightly mediate chemoresistance (10), which has been demonstrated in various
cancer types, including lung (11),
pancreatic (12), prostate (13), liver (14) and head and neck squamous cancer
(15). Yin et al (16) reported that enrichment of CSCs in the
Panc-1 pancreatic cancer cell line increased the migration ability
and resistance to gemcitabine, although the mechanism has remained
to be elucidated.
Long non-coding RNAs (lncRNAs) are a class of
endogenous cellular RNAs of <200 nucleotides in length that lack
an open reading frame of significant length (17). In recent years, accumulating evidence
has indicated regulatory roles of lncRNAs regarding the malignant
character of various cancer types. Overexpression of
metastasis-associated lung adenocarcinoma transcript 1 (MALAT-1), a
highly evolutionarily conserved and ubiquitously expressed lncRNA,
in pancreatic cancer cells increased the proportion of pancreatic
CSCs, maintained their self-renewal capacity, decreased their
sensitivity to anticancer drugs and accelerated tumor angiogenesis
in vitro (18). HOX
transcript antisense RNA (HOTAIR) has been intensely investigated
in several cancer types, including lung (19), prostate (20) and pancreatic cancers (21). HOTAIR and MALAT-1 have received
increasing attention due to their aberrant expression in cancer
tissues (18–21).
In the present study, the CSC sub-population from
Panc-1 cells was enriched using serum-free medium and exposed to
different concentration of gemcitabine. The cells were then
subjected to reverse-transcription quantitative polymerase chain
reaction (RT-qPCR) analysis to detect the expression levels of
several lncRNAs. Apart from HOTAIR and MALAT-1 which were assessed
due to their tight association with the malignancy of pancreatic
cancer, maternally expressed 3 (MEG3) (21), protein phosphatase 3 catalytic
subunit β (PPP3CB), mitogen-activated protein kinase kinase kinase
14 (MAP3K14) and death-associated protein kinase 1 (DAPK1)
(22) were also assessed. A
significantly higher expression of HOTAIR was observed in Panc-1
and CSCs enriched from Panc-1 after exposure to gemcitabine.
Further experiments strongly suggested that HOTAIR may have a role
in pancreatic stemness, increasing chemoresistance to gemcitabine,
attenuating apoptosis and promoting proliferation. Taken together,
these results provided novel insight into the negative effects of
gemcitabine exposure on the sub-population of pancreatic CSCs by
upregulating HOTAIR and uncovered a role for the lncRNA HOTAIR as a
potential stemness regulator and novel therapeutic target.
Materials and methods
Cell culture and gemcitabine
treatment
The Panc-1 pancreatic cancer cell line purchased
from American Type Culture Collection (ATCC; Manassas, VA, USA) and
cultured in Dulbecco's Modified Eagle's Medium (DMEM; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
penicillin (100 U/ml) and streptomycin (100 U/ml; Thermo Fisher
Scientific, Inc.) at 37°C in an incubator with 5% CO2.
To stimulate pancreatic cancer cells to form tumor spheres in
suspension, the following culture conditions were used: Panc-1
cells were suspended using Trypsin (Thermo Fisher Scientific, Inc.
CA, USA) and diluted to a density of 106 cells/ml in
serum-free medium, which was composed of DMEM/F12 supplemented with
2% B-27 (Thermo Fisher Scientific, Inc.), 20 ng/ml epidermal growth
factor (EGF) and 10 ng/ml fibroblast growth factor-basic (bFGF;
PeproTech, Rocky Hill, NJ, USA). The cells were passaged every 12
days and replated in the serum-free medium. The spheres forming
under these conditions were named PANC-1 CSCs.
For gemcitabine treatment, cells were cultured in
SFM supplemented with 25, 50 or 100 µg/ml gemcitabine
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 24 h, and then
subjected to the other assays.
Construction of lentiviral particles
containing HOTAIR coding sequence
The HOTAIR coding sequence was amplified by RT-PCR
and then cloned into the pCDH-MSCV-mcs-GFP lentiviral vector
(System Biosciences; Palo Alto, CA, USA) at the EcoRI and NotI
sites (Fermentas; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). For generating lentiviral particles, the packaging vectors
psPAX2 and pMD2.G (Addgene, Inc., Cambridge, MA, USA) were
co-transfected into 293T cells (ATCC) with pCDH-MSCV-msc-GFP
lentiviral vector containing HOTAIR coding sequence. After 72 h,
the supernatant was collected and the titer was determined.
Construction of the small hairpin RNA
targeting HOTAIR (shHOTAIR) vector and plasmid transfection
For constructing the vector encoding shHOTAIR, the
following specific oligonucleotides targeting HOTAIR were
synthesized (Shenggong, Shanghai, China):
Sense,5′-GATCCGCCACATGAACGCCCAGAGATTTTCAAGAGAAATCTCTGGGCGTTCATGTGGTTTTTTG-3′
and
anti-sense,5′-AATTCAAAAAACCACATGAACGCCCAGAGATTTCTCTTGAAAATCTCTGGGCGTTCATGTGGCG-3′.
After annealing, the double-stranded DNA was inserted into
pENTR.hU6hH1 empty vector between NdeI and EcoRI
sites (Fermentas; Thermo Fisher Scientific, Inc.). The
pENTR-shHOTAIR plasmids or empty vectors were transfected into
target cells using Lipofectamine 2000™ (Thermo Fisher Scientific,
Inc.).
RT-qPCR
Total RNA extracted from target cells by using
TRIzol reagent (Thermo Fisher Scientific, Inc.) was used for
complementary (c)DNA synthesis. Briefly, M-MLV first strand cDNA
Synthesis kit (Omega Bio-Tek, Inc., Norcross, GA, USA) was used
following manufacturer's guide. The sequences of the primers used
in the present study were as follows: HOTAIR forward,
5′-GAGAGAGGGAGCCCAGAGTT-3′ and reverse, 5′-GCTTGGGTGTAATTGCTGGT-3′;
MALAT-1 forward, 5′-TGTGTGCCAATGTTTCGTTT-3′ and reverse,
5′-AGGAGAAAGTGCCATGGTTG-3′; MEG3 forward,
5′-TTGACAGGTCAGTCCCTTCC-3′ and reverse, 5′-TTCCACGGAGTAGAGCGAGT-3′;
PPP3B forward, 5′-CAACCATGAATGCAGACACC-3′ and reverse,
5′-TGGTGAAAGTCCACCATGAA-3′; MAP3K14 forward,
5′-CAAGCCTCTGAAGGAACCAG-3′ and reverse, 5′-AGGGATGAGGCAGTCTGCTA-3′;
DAPK1 forward, 5′-ATGATCCCACGTCAATCCAT-3′ and reverse,
5′-CCACCAGGACAACTTGGAGT-3′; GAPDH forward,
5′-GGAGCGAGATCCCTCCAAAAT-3′ and reverse,
5′-GGCTGTTGTCATACTTCTCATGG-3′. PCR was performed on an Applied
Biosystems 7500 Real-time system (Thermo Fisher Scientific, Inc.)
using Power SYBR-Green PCR Master Mix (Thermo Fisher Scientific,
Inc.). The thermocycling conditions were as follows: 5 min at 50°C
and 5 min at 95°C, followed by 35 cycles of 30 sec at 95°C and 60
sec at 60°C. Experiments were performed in triplicate using the
2−ΔΔCq method (23).
Transwell migration assay
The Transwell migration assay was performed using a
24-well Transwell chemotaxis chamber (EMD Millipore, Billerica, MA,
USA). In brief, DMEM/F12 (500 µl) supplemented with 2% B-27, 10
ng/ml EGF, and 20 ng/ml bFGF was placed in the lower chamber. A
total of 2×104 CSCs in a single-cell suspension in 200
µl medium were seeded into the upper chamber (membrane pore size, 8
µm). The chamber was then incubated for 24 h at 37°C in a
humidified atmosphere with 5% CO2. The membrane was
removed and cells on the upper surface which had not migrated were
wiped away with a cotton swab. Subsequently, the membrane was fixed
in 4% paraformaldehyde for 5 min at room temperature and then
stained with 0.1% crystal violet (Sigma-Aldrich; Merck KGaA) for 10
min, followed by 3 washes with ice-cold PBS. The number of cells
that had migrated to the lower surface of the membrane was counted
in 10 random high-power fields under a light microscope (BL-AC10DS;
Olympus, Tokyo, Japan). Each assay was performed in triplicate
wells.
Serial replating experiments
Target cells were transfected with shHOTAIR or with
LV-HOTAIR for 48 h. For serial replating experiments, cells were
replated at a clonal density (1,000 cells/well) and cultured in
serum-free medium supplemented with 2% B-27, 10 ng/ml EGF and 20
ng/ml bFGF. Every 3 days, the medium was half-replaced. After 14
days, cells were washed with PBS, fixed with 4% paraformaldehyde in
PBS, stained with 0.1% crystal violet for 10 min and washed again
with PBS, and the colonies were counted. For replating, the same
amount of cells was plated in serum-free medium. After 14 days, the
same procedure was performed three times.
Cell Counting kit-8 (CKK-8)
proliferation assay
Cells at a concentration of 5×103 were
seeded into 24-well culture plates in 500 µl culture medium
supplemented with 2% B-27, 10 ng/ml EGF and 20 ng/ml bFGF. Prior to
detection, CCK-8 reagent (Sigma-Aldrich; Merck KGaA) was added at
10 µg/well, followed by incubation for 2–4 h at 37°C and 5%
CO2 according to the manufacturer's protocols. A cell
growth curve was drawn based on the corresponding normalized
optical density values at 450 nm and each data-point represents the
mean of three independent samples.
Flow cytometric analysis
For flow cytometric analysis of CSC markers, cells
were detached using 0.25% Trypsin, re-suspended at 106
cells/ml and incubated with anti-CD24-fluorescein isothiocyanate
(FITC; cat. no. FCMAB188F) and anti-CD44-phycoerythrin (cat. no.
MABF582; EMD Millipore, Billerica, MA, USA) according to the
manufacturer's protocols for 30 min on ice. Following washing with
PBS three times, cells were fixed with 4% paraformaldehyde and then
subjected to flow cytometric analysis.
Apoptosis was detected by flow cytometry following
double staining with Annexin V-FITC and propidium iodide using the
Annexin V-FITC Apoptosis Detection kit (BD Biosciences- Franklin
Lakes, NJ, USA). A total of 0.5 ml (1×106 cells/ml) of
treated cells were washed in PBS, re-suspended in binding buffer
supplied in the kit and stained with FITC-conjugated Annexin V (BD
Pharmingen; BD Biosciences). After being stained for 30 min at 4°C,
the cells were incubated for 15 min in the dark at room
temperature. Cells were re-washed with binding buffer and analysed
using a flow cytometr (BD FACS Canto II; BD Biosciences).
Statistical analysis
Data were expressed as the mean ± standard
deviation. Multigroup comparisons of the mean were performed by
one-way analysis and Specific contrasts were generated by Tukey's
post hoc comparisons. using SPSS 16.0 software package (SPSS, Inc.,
Chicago, IL, USA. P<0.05 was considered to indicate a
statistically significant difference.
Results
Short-term exposure to gemcitabine
induces expression of HOTAIR in PANC-1 CSCs
For detecting the expression profile of HOTAIR,
MALAT-1, MEG3, PPP3CB, MAP3K14 and DAPK1 in PANC-1 and PANC-1 CSCs,
CSCs were enriched from the PANC-1 population by incubation in
serum-free medium. The self-renewal capacity of enriched CSCs was
analyzed by a serial replating assay and the results confirmed
their self-renewal capacity (Fig.
1A), while Panc-1 cells failed to form countable spheres
because of its weak clonogenicity (data not shown). Detection of
the population of CD24−/CD44+ cells, which is
the CSC population, also revealed a high enrichment compared with
native PANC cells (Fig. 1B). The
Panc-1 cells and the enriched Panc-1 CSCs with or without
gemcitabine exposure were then subjected to RT-qPCR analysis. In
Panc-1 cells, compared with untreated cells, 2 µM gemcitabine
exposure significantly upregulated MALAT-1 (Fig. 1C), and in Panc-1 CSCs, 2 µM
gemcitabine upregulated HOTAIR and MALAT-1 (Fig. 1D). As gemcitabine exposure did not
affect HOTAIR in Panc-1 cells, the subsequent experiments focused
on the regulatory roles of HOTAIR on Panc-1 CSCs.
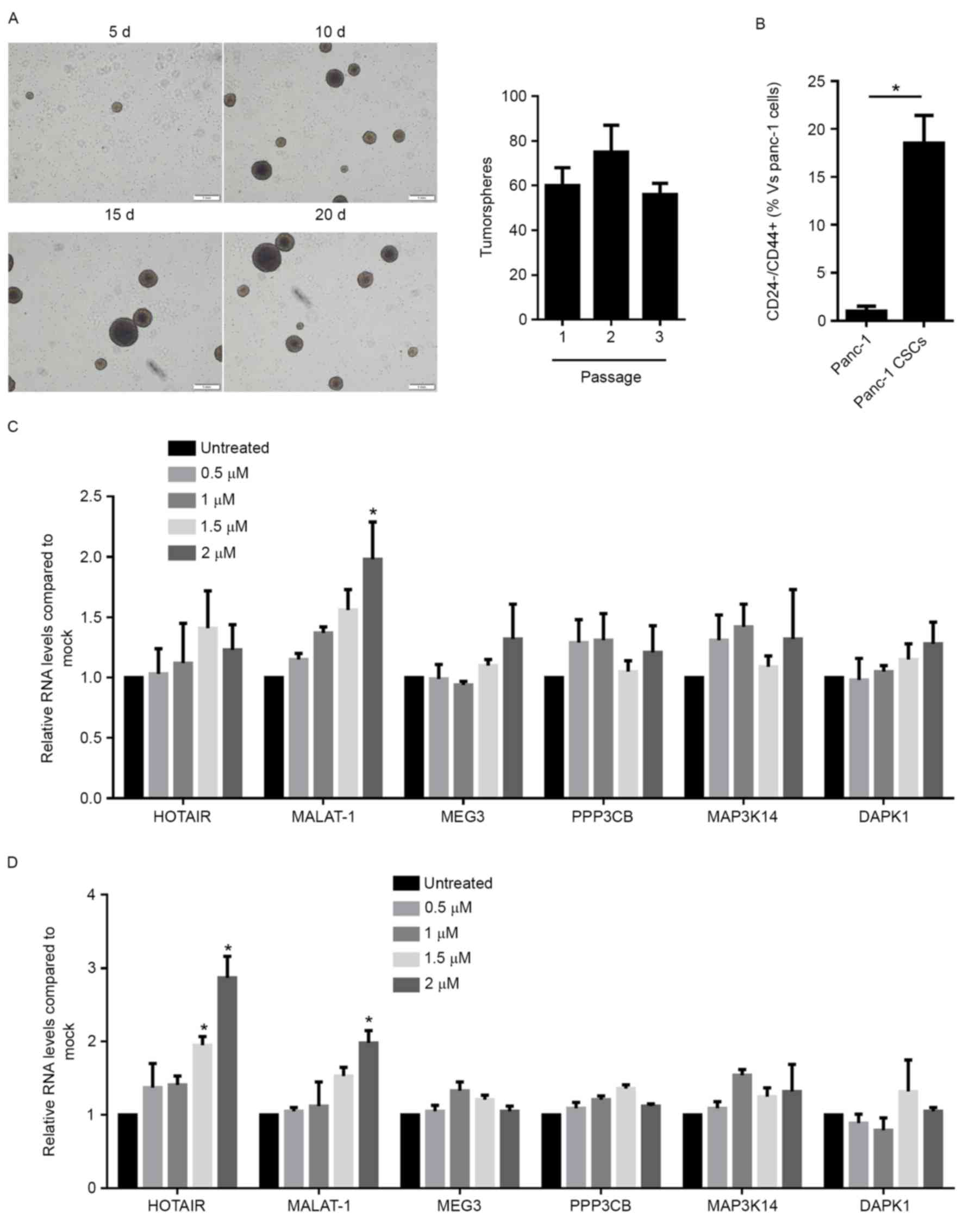 | Figure 1.Gemcitabine exposure leads to
upregulation of long non-coding RNA HOTAIR expression in Panc-1
CSCs. (A) Enrichment of CSCs from Panc-1 cells and identification
of their self-renewal capacity by serial replating assay (scale
bar, 1 mm). (B) Flow cytometric analysis of
CD24−/CD44+ cells in Panc-1 cells and Panc-1
CSCs. The amount of CD24−/CD44+ cells in
Panc-1 cells was considered as 1. Reverse-transcription
quantitative polymerase chain reaction analysis of the expression
levels of HOTAIR, MALAT-1, MEG3, PPP3CB, MAP3K14 and DAPK1 in (C)
Panc-1 cells or (D) Panc-1 CSCs with or without gemcitabine
exposure. *P<0.05 vs. untreated or as indicated. CSCs, cancer
stem-like cells; HOTAIR, HOX antisense intergenic RNA; MALAT-1,
metastasis associated lung adenocarcinoma transcript 1; MEG3,
maternally expressed 3; PPP3CB, protein phosphatase 3 catalytic
subunit β; MAP3K14, mitogen-activated protein kinase kinase kinase
14; DAPK1, death-associated protein kinase 1; d, days. |
Overexpression of HOTAIR increases
chemoresistance to gemcitabine in PANC-1 CSCs
The upregulation of HOTAIR after gemcitabine
treatment in CSCs prompted us to investigate the potential effects
of HOTAIR on the chemoresistance of PANC-1 CSCs. PANC-1 CSCs were
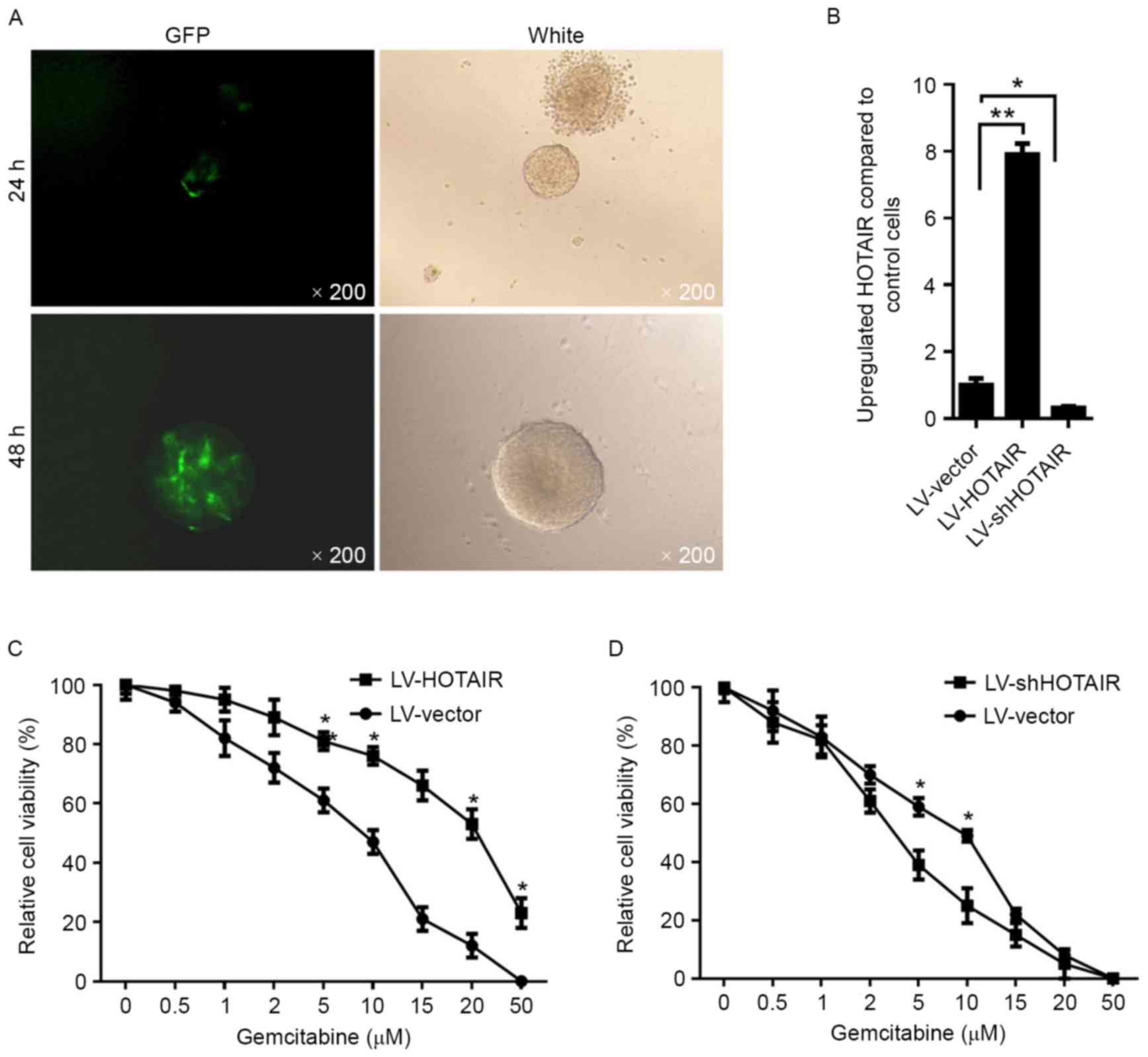
transfected by a HOTAIR-expressing lentivirus containing an GFP
coding sequence for 48 h. Subsequently, PANC-1 CSCs were imaged by
fluorescent microscopy, revealing high efficiency of lentiviral
transfection (Fig. 2A). For
introducing HOTAIR into PANC-1 CSCs, lentivirus containing a coding
sequence for HOTAIR (LV-HOTAIR) was packaged. At 48 h after
transfection with LV-HOTAIR, the overexpression of HOTAIR compared
with that in PANC-1 CSCs transfected with empty LV vector was
confirmed by RT-qPCR (Fig. 2B).
Subsequently, the sensitivity to gemcitabine was assessed,
indicating that overexpression of HOTAIR significantly decreased
the sensitivity of PANC-1 CSCs to gemcitabine (Fig. 2C). To further confirm the effect of
HOTAIR on gemcitabine resistance, siHOTAIR was transfected into
PANC-1 CSCs. Following knockdown of HOTAIR (Fig. 2B), the sensitivity of PANC-1 CSCs to
gemcitabine was enhanced (Fig.
2E).
Overexpression of HOTAIR attenuates
apoptosis and promotes proliferation of PANC-1 CSCs under
gemcitabine treatment
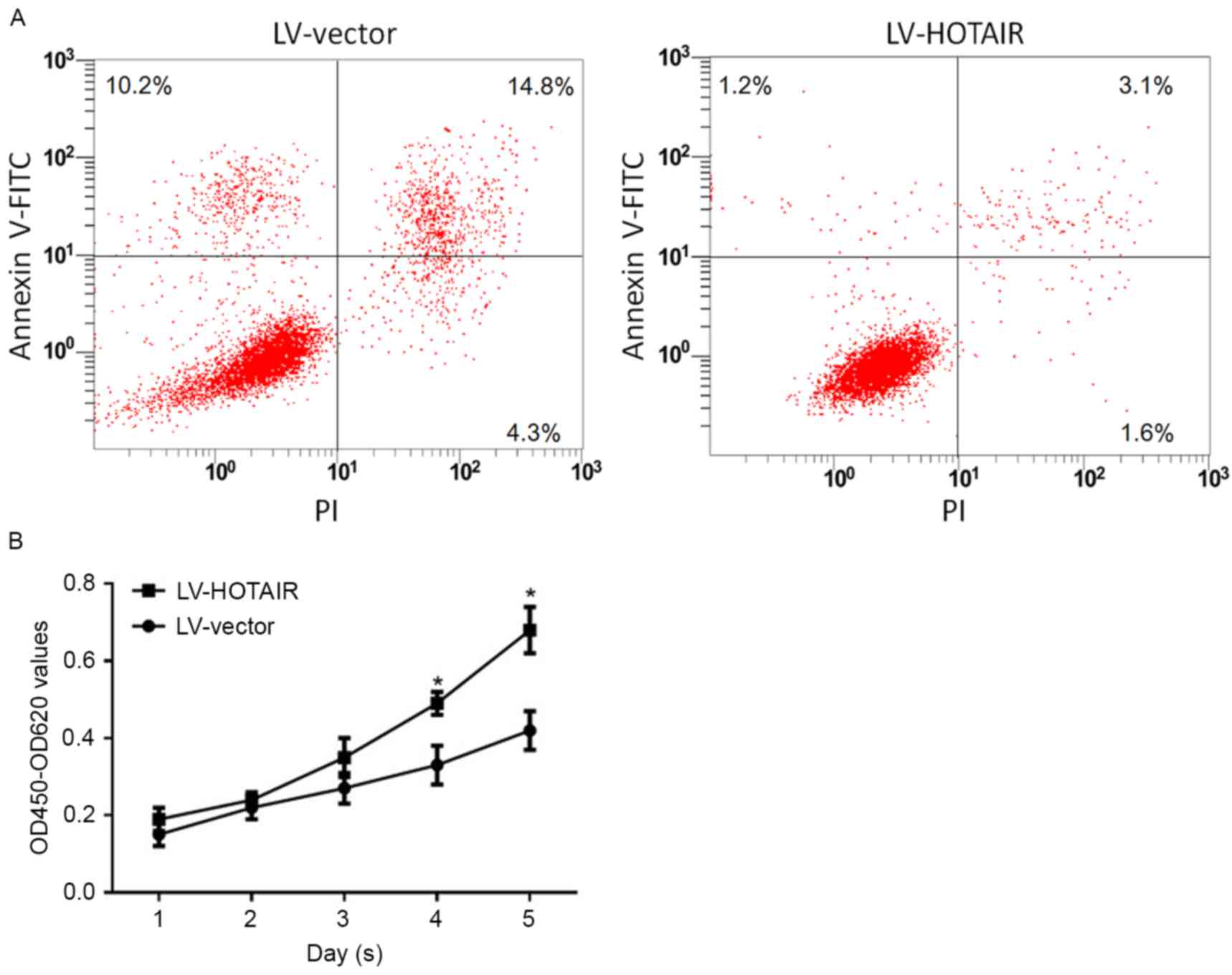
According to the above results (Fig. 2), upregulation of HOTAIR in PANC-1
CSCs increased the concentration leading to 30% inhibition (IC30)
and IC50 of gemcitabine. This prompted us to assess whether the
resistance effect of HOTAIR attenuates apoptosis and promotes the
proliferation of PANC-1 CSCs. After treatment with gemcitabine at
the IC50 concentration for 24 h, Annexin V/propidium iodide (PI)
double labeling was performed for analyzing the apoptotic rate. The
results indicated that upregulation of HOTAIR, but not transfection
with empty LV vector, decreased the ratio of early apoptotic cells
(Annexin V-FITC+ and PI− due to intact cell
membrane) and late apoptotic cells (Annexin V-FITC+ and
PI+ due to perforated cell membrane) (Fig. 3A). In comparison with untransfected
cells, no detectable difference was observed in cells transfected
with empty LV vector. Of note, the population of necrotic cells
(Annexin V-FITC−/PI+) exhibited a slight
change (Fig. 3A). For detecting the
effect of HOTAIR on the proliferation capacity, 1×104
transfected PANC-1 CSCs were incubated with the IC30 concentration
of gemcitabine for 24–96 h, and the cellular viability was detected
on each day. The results indicated that upregulation of HOTAIR
promoted the proliferation of PANC-1 CSCs under gemcitabine
treatment (Fig. 3B). Surprisingly,
knockdown of HOTAIR by shHOTAIR introduction failed to
significantly affect apoptosis or proliferation (data not shown),
possibly due to the low expression levels of HOTAIR in unstressed
PANC-1 CSCs.
Upregulation of HOTAIR affects the
self-renewal capacity, migration and colony formation capacities of
PANC-1 CSCs
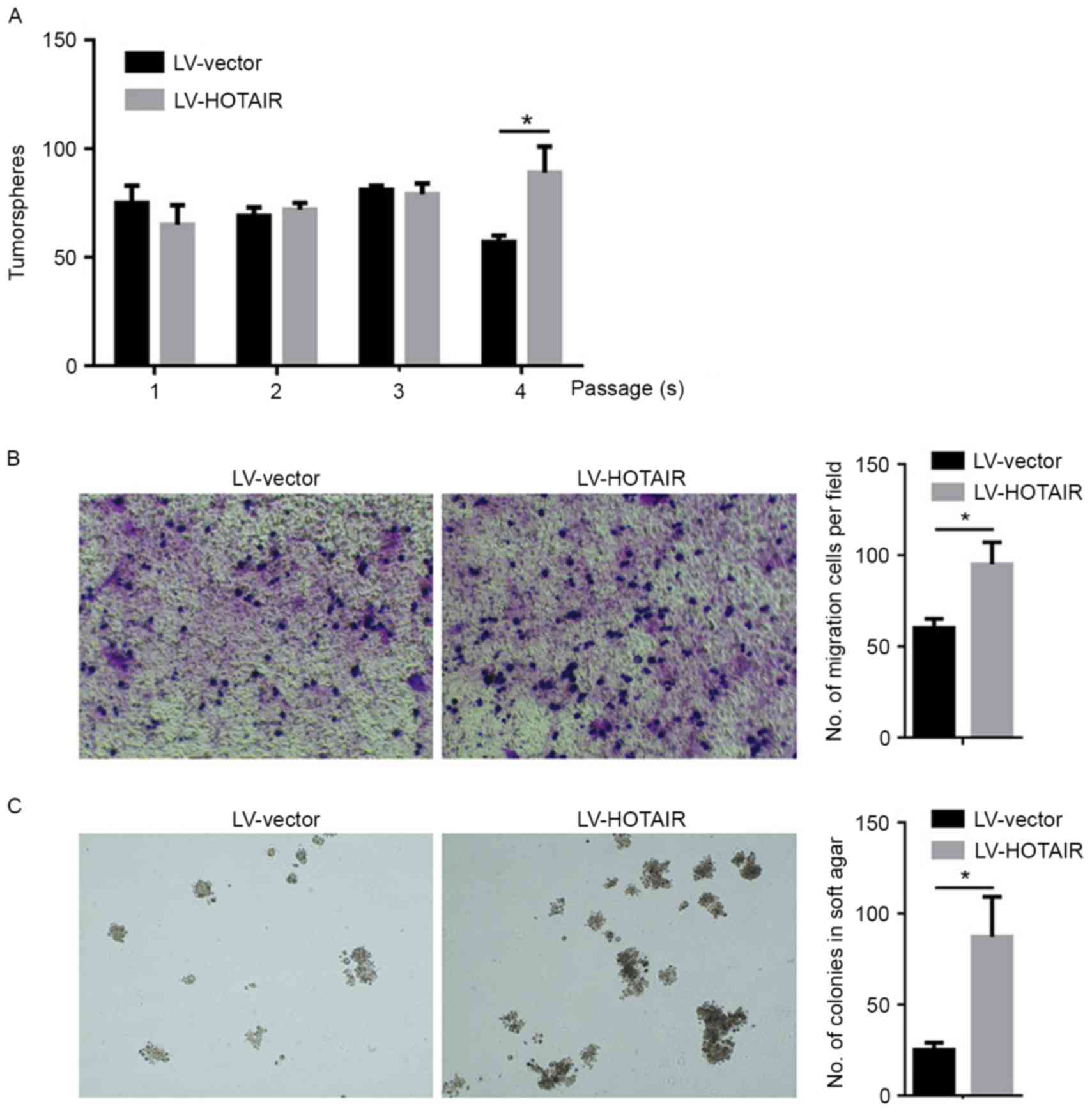
The present study further investigated the
regulatory roles of upregulated HOTAIR in biological processes of
PANC-1 CSCs. To analyse the impact of HOTAIR on the self-renewal
capacity of PANC-1 CSCs, a well-established serial replating assay
was used. LV-HOTAIR-transfected PANC-1 CSCs were able to form
colonies in all four rounds of replating, and exhibited no
significant difference in the initial three rounds of replating
(Fig. 4A). However, the empty
LV-vector-transfected PANC-1 CSCs exhibited a significant decrease
in their self-renewal capacity during the 4th round of replating
(Fig. 4A). A Transwell-based
migration assay was established to quantitatively evaluate PANC-1
CSCs migration in vitro. As presented in Fig. 4B, compared with the control group,
the average number of migrated PANC-1 CSCs increased significantly
after HOTAIR introduction. To examine the effects of HOTAIR on
colony formation in PANC-1 CSCs, a colony formation assay on soft
agar was performed. The number of colonies formed by
LV-HOTAIR-transfected PANC-1 CSCs was significantly increased
compared with that of empty vector-transfected PANC-1 CSCs
(Fig. 4C).
Discussion
The present study demonstrated that lncRNA HOTAIR
was induced in Panc-1 CSCs after short-term gemcitabine exposure.
Several lncRNAs tightly associated with malignancy of pancreatic
cancer, including MALAT-1, HOTAIR, MEG3, PPP3CB, MAP3K14 and DAPK1
were detected in Panc-1 and Panc-1 CSCs, revealing that HOTAIR was
investigated as its upregulation was CSC-specific. This prompted us
to focus on the regulatory effects of HOTAIR induced by gemcitabine
on the self-renewal capacity, proliferation, apoptosis and
migration of Panc-1 CSCs. As expected, induction of HOTAIR by
gemcitabine treatment promoted the proliferation and migration,
maintained the self-renewal capacity and attenuated apoptosis of
Panc-1 CSCs. Of note, following gemcitabine treatment for a
relative long duration (96 h), HOTAIR expression was not
significantly changed compared with that in untreated Panc-1 CSCs
(data not shown). These results indicated that the induction of
HOTAIR after gemcitabine exposure may be antagonized in an unknown
manner. Taken together, induction of HOTAIR by short-term exposure
to gemcitabine may contribute to the chemoresistance of Panc-1
CSCs. Furthermore, upregulation of HOTAIR led to the promotion of
the proliferation and migration, maintenance of the self-renewal
capacity and inhibition of apoptosis of Panc-1 CSCs after treatment
with gemcitabine.
According to the CSC hypothesis, a small
sub-population within a tumor has multipotent features and the
capacity for indefinite self-renewal and asymmetric cell division
(24,25). Not only in carcinogenesis,
accumulating evidence has indicated that CSCs may have a critical
role in cancer aggressiveness, metastasis, recurrence and
chemoresistance of solid tumors including pancreatic adenocarcinoma
(26). CSCs were reported to be
tightly associated with increased chemoresistance of pancreatic
cancer with several mechanisms. Cioffi et al (27) found that, in pancreatic CSCs,
downregulation of the microRNA-17-92 cluster promoted the
self-renewal capacity as well as the in vivo tumorigenicity
and chemoresistance by targeting multiple members of the
Nodal/activin/transforming growth factor-β1 signalling cascade. The
enhanced efflux of Hoechst 33342 dye through adenosine triphosphate
binding cassette transporters by CSCs demonstrated their
chemoresistance mechanism through elimination of drug molecules
(28). Furthermore, aldehyde
dehydrogenase 1, a potential marker for CSCs, has been identified
to have a potential role in chemoresistance (29). The role of B-cell lymphoma-2 (Bcl-2)
protein and its family members has also been well explored as a
novel mechanism of chemoresistance in CSCs (30). Collectively, CSCs of pancreatic
cancer cells, contribute to chemoresistance via a variety of
mechanisms.
The present study aimed to investigate the
association and potential role of lncRNAs with the chemoresistant
capacity of pancreatic CSCs, as emerging evidence has demonstrated
the critical roles of lncRNAs in inducing chemoresistance in
several cancer types. Li et al (31) reported that, in nasopharyngeal
carcinoma (NPC), the recently identified lncRNA ROR is associated
with the proliferation, metastasis, apoptosis and chemoresistance
of NPC. MEG3 was revealed to be partially responsible for
regulating cisplatin resistance of human lung adenocarcinoma cells
through control of p53 and Bcl extra large protein expression
(32). Of note, it was also reported
that changes in the expression of ncRNAs may be associated with
chemoresistance of non-small-cell lung cancer cells (33). As expected, HOTAIR was found to be
induced by gemcitabine exposure and the ectopic expression of
HOTAIR led to the promotion of proliferation and migration as well
as maintenance of the self-renewal capacity of pancreatic CSCs.
In conclusion, the present study was the first, to
the best of our knowledge, to demonstrate that lncRNA HOTAIR is
induced by gemcitabine in pancreatic CSCs, and induction of HOTAIR
expression led to promotion of proliferation and migration,
maintenance of self-renewal capacity, attenuation of apoptosis and
increase of chemoresistance. However, the exact mechanisms by which
HOTAIR regulates these processes requires further elucidation.
Based on these data, further study of the effects of HOTAIR on
pancreatic CSCs is required in pathological tissues rather than a
cell line. In addition, the regulation of associated genes and
protein functions should also be studied. These further studies
will help to improve the clinical treatment of pancreatic
cancer.
Acknowledgements
The present study was supported by the Sichuan
Provincial Scientific Grant (grant no. 2012SZ0141).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Reilly EM and Abou-Alfa GK: Cytotoxic
therapy for advanced pancreatic adenocarcinoma. Semin Oncol.
34:347–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Z, Li Y, Ahmad A, Banerjee S, Azmi
AS, Kong D and Sarkar FH: Pancreatic cancer: Understanding and
overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 8:27–33.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bergman AM, Pinedo HM and Peters GJ:
Determinants of resistance to 2′,2′-difluorodeoxycytidine
(gemcitabine). Drug Resist Updat. 5:19–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andersson R, Aho U, Nilsson BI, Peters GJ,
Pastor-Anglada M, Rasch W and Sandvold ML: Gemcitabine
chemoresistance in pancreatic cancer: Molecular mechanisms and
potential solutions. Scand J Gastroenterol. 44:782–786. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Polyak K and Hahn WC: Roots and stems:
Stem cells in cancer. Nat Med. 12:296–300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kakarala M and Wicha MS: Implications of
the cancer stem-cell hypothesis for breast cancer prevention and
therapy. J Clin Oncol. 26:2813–2820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:5821–5828.
2003.PubMed/NCBI
|
|
10
|
Abdullah LN and Chow EK: Mechanisms of
chemoresistance in cancer stem cells. Clin Transl Med. 2:32013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ucar D, Cogle CR, Zucali JR, Ostmark B,
Scott EW, Zori R, Gray BA and Moreb JS: Aldehyde dehydrogenase
activity as a functional marker for lung cancer. Chem Biol
Interact. 178:48–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jimeno A, Feldmann G, Suárez-Gauthier A,
Rasheed Z, Solomon A, Zou GM, Rubio-Viqueira B, García-García E,
López-Ríos F, Matsui W, et al: A direct pancreatic cancer xenograft
model as a platform for cancer stem cell therapeutic development.
Mol Cancer Ther. 8:310–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hellsten R, Johansson M, Dahlman A,
Sterner O and Bjartell A: Galiellalactone inhibits stem cell-like
ALDH-positive prostate cancer cells. PLoS One. 6:e221182011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma S, Chan KW, Lee TK, Tang KH, Wo JY,
Zheng BJ and Guan XY: Aldehyde dehydrogenase discriminates the
CD133 liver cancer stem cell populations. Mol Cancer Res.
6:1146–1153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Clay MR, Tabor M, Owen JH, Carey TE,
Bradford CR, Wolf GT, Wicha MS and Prince ME: Single-marker
identification of head and neck squamous cell carcinoma cancer stem
cells with aldehyde dehydrogenase. Head Neck. 32:1195–1201. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin T, Wei H, Gou S, Shi P, Yang Z, Zhao G
and Wang C: Cancer stem-like cells enriched in Panc-1 spheres
possess increased migration ability and resistance to gemcitabine.
Int J Mol Sci. 12:1595–1604. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta. 1839:1097–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiao F, Hu H, Han T, Yuan C and Wang L,
Jin Z, Guo Z and Wang L: Long noncoding RNA MALAT-1 enhances stem
cell-like phenotypes in pancreatic cancer cells. Int J Mol Sci.
16:6677–6693. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuang Y, Wang X, Nguyen HT, Zhuo Y, Cui
X, Fewell C, Flemington EK and Shan B: Induction of long intergenic
non-coding RNA HOTAIR in lung cancer cells by type I collagen. J
Hematol Oncol. 6:352013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang A, Zhao JC, Kim J, Fong KW, Yang YA,
Chakravarti D, Mo YY and Yu J: LncRNA HOTAIR enhances the
androgen-receptor-mediated transcriptional program and drives
castration-resistant prostate cancer. Cell Rep. 13:209–221. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim K, Jutooru I, Chadalapaka G, Johnson
G, Frank J, Burghardt R, Kim S and Safe S: HOTAIR is a negative
prognostic factor and exhibits pro-oncogenic activity in pancreatic
cancer. Oncogene. 32:1616–1625. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tahira AC, Kubrusly MS, Faria MF, Dazzani
B, Fonseca RS, Maracaja-Coutinho V, Verjovski-Almeida S, Machado MC
and Reis EM: Long noncoding intronic RNAs are differentially
expressed in primary and metastatic pancreatic cancer. Mol Cancer.
10:1412011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee CJ, Dosch J and Simeone DM: Pancreatic
cancer stem cells. J Clin Oncol. 26:2806–2812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hermann PC, Mueller MT and Heeschen C:
Pancreatic cancer stem cells-insights and perspectives. Expert Opin
Biol Ther. 9:1271–1278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Habib M and Saif MW: Pancreatic cancer
stem cells: Their role in pancreatic cancer patient outcomes and
what is future? JOP. 14:401–404. 2013.PubMed/NCBI
|
|
27
|
Cioffi M, Trabulo SM, Sanchez-Ripoll Y,
Miranda-Lorenzo I, Lonarod E, Dorado J, Vieira Reis C, Ramirez JC,
Hidalgo M, Aicher A, et al: The miR-17-92 cluster counteracts
quiescence and chemoresistance in a distinct subpopulation of
pancreatic cancer stem cell. Gut. 64:1936–1948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goodell MA, Brose K, Paradis G, Conner AS
and Mulligan RC: Isolation and functional properties of murine
hematopoietic stem cells that are replicating in vivo. J Exp Med.
183:1797–1806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hilton J: Role of aldehyde dehydrogenase
in cyclophosphamide-resistant L1210 leukemia. Cancer Res.
44:5156–5160. 1984.PubMed/NCBI
|
|
30
|
Kim R, Emi M and Tanabe K: Role of
mitochondria as the gardens of cell death. Cancer Chemother
Pharmacol. 57:545–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li L, Gu M, You B, Shi S, Shan Y, Bao L
and You Y: Long non-coding RNA ROR promotes proliferation,
migration and chemoresistance of nasopharyngeal carcinoma. Cancer
Sci. 107:1215–1222. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Wan L, Lu K, Sun M, Pan X, Zhang P,
Lu B, Liu G and Wang Z: The long noncoding RNA MEG3 contributes to
cisplatin resistance of human lung adenocarcinoma. PLoS One.
10:e01145862015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Li H, Hou S, Hu B, Liu J and Wang
J: The noncoding RNA expression profile and the effect of
lncRNAAK126698 on cisplatin resistance in non-small-cell lung
cancer cell. PLoS One. 8:e653092013. View Article : Google Scholar : PubMed/NCBI
|