Introduction
Osteoarthritis (OA) is the most common arthritis
characterized by a hallmark symptom of pain, even leads to
disability (1). It is well-known
that OA is caused by the combined effects of genetic, biological,
and biomechanical factors (2). With
the rise of obesity as well as an ageing population, the prevalence
of OA is increasing (2). The quality
of life of these population is poor in the end-stage of OA due to
joint destruction (3). Currently,
joint replacement is considered as an effective treatment for
end-stage OA; however, high socioeconomic cost, poor functional
outcomes and the limited lifespan of prostheses influence its
application and efficacy (4,5). Thus, it is imperative to search for new
and effective intervention and therapy for OA.
Recently, microRNAs (miRNAs/miRs), small non-coding
RNAs, play a significant regulatory role in oncogenesis through
mediating target genes expressions (6). Increasing evidences suggest that miRNAs
are widely involved in immune response, inflammation reaction,
infection as well as cell metabolism, growth and migration
(7,8). Accumulating studies have focused on the
potential roles of miRNAs in various diseases, such as autoimmune
diseases (9), cardiovascular
diseases (10), neurodegeneration
diseases (11), and cancers
(12). Thus, understanding the
mechanisms of miRNAs in OA development may contribute to search for
an effective treatment for this disease.
It has been shown that miR-4262 exerts the vital
roles in a variety of tissues and cells. Previous study has
suggested that miR-4262 participates in the development of acute
lung injury through regulating the apoptosis of pulmonary
endothelial cells (13). Zhang et
al (14) and Lu et al
(15) have demonstrated that
miR-4262 exerts pro-proliferation effect in human cutaneous
malignant melanoma cells and hepatocellular carcinoma cells. In
addition, up-regulated miR-4262 can inhibit osteopontin-mediated
cell invasion in osteosarcoma (16).
Although the pathogenic effects of miR-4262 on several diseases
have been disclosed, the role of miR-4262 in OA is still
unclear.
Sirtuin type 1 (SIRT1), a mammalian homolog of
silent information regulator 2 (Sir2), has been found to be a key
regulator in the pathogenesis of OA (17). In chondrocytes, SIRT1 reduction may
result in chondrocyte hypertrophy and cartilage matrix loss
(17). Moreover, inhibition of SIRT1
induces chondrocyte apoptosis via modulating mitochondria-related
apoptotic signals (18). It is also
reported that disruption of SIRT1 in chondrocytes can accelerate OA
progression under mechanical stress and during ageing in mice
(19). Furthermore, activation SIRT1
can protect chondrocytes from tumor necrosis factor-α
(TNF-α)-induced inflammatory effects, and SIRT1 activators may be
explored as potential treatments for OA (20). Given the key role of SIRT1 in OA, we
hypothesized that miR-4262 may play a key role in OA development
via regulating SIRT1. In the present study, primary chondrocytes
were separated, and OA cell model was induced by the treatment of
TNF-α. The level of miR-4262 was detected in TNF-α-treated
chondrocytes, and then the effects of aberrant expression of
miR-4262 or its target gene SIRT1 on cell viability, cell
apoptosis, cell autophagy, and matrix synthesis, as well as the
expressions of proteins related to phosphoinositide 3-kinase
(PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway
were evaluated, aiming to investigate the effect and underlying
mechanism of miR-4262 in OA.
Materials and methods
Isolation and culture of primary
articular chondrocytes
Approval from the Ethics Committee of Animal
Experimental Center of University, and all experiments were
performed following the guidelines of the Institutional Animal Care
and Use Committee. Six healthy Sprague-Dawley (SD) rats (200–250 g)
were provided to isolate primary articular chondrocytes as
previously described (21). In
brief, the cartilage tissues from knee joints of rats were sliced
into small pieces, and then digested with DMEM/F12 medium
containing 100 U/ml penicillin, 100 µg/ml streptomycin, 10% fetal
bovine serum (FBS), and 0.2% Type II collagenase for 24 h at 37°C.
Undigested cartilages were removed and primary chondrocytes were
maintained in DMEM/F12 medium. The isolated chondrocytes were
confirmed by the histological observation, and differentiation
markers in chondrocytes, including collagen type 2 (Col2) and
aggrecan, was detected by quantitative PCR (qPCR) analysis. OA
model of chondrocytes was then induced by 20 ng/ml of TNF-α
(PeproTech, Rocky Hill, NJ, USA). To evaluate the role of miR-4262
in OA, OA-chondrocytes were transfected with scramble (control of
mimic), miR-4262 mimic, NC (control of inhibitor), miR-4262
inhibitor, pEX (control of pEX-SIRT1), pEX-SIRT1, siNC (control of
si-SIRT1), or si-SIRT1 using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cells with various treatments were exposed to 2 µM rapamycin (RAPA,
autophagic activator) to evaluate the cell autophagy. In addition,
we performed similar autophagy tests in the presence or absence of
lysosomal protease inhibitors E64d (1 mg/ml; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and pepstatin A (1 mg/ml; Sigma-Aldrich;
Merck KGaA).
Cell viability assay
Chondrocytes (5×103 cells/well) were
grown in 96-well plates and received the above various treatments
for 0, 12, 24, 48 h. Cell Counting Kit-8 (CCK-8; Dojindo
Laboratory, Kumamoto, Japan) was used to detect cell viability.
Cells were treated with 10% WST-8 for 4 h at 37°C, then the
absorbance at 450 nm was measured using a microplate reader
(Molecular Devices, Sunnyvale, CA, USA).
TUNEL staining
After various treatments, cells apoptosis was
performed using TUNEL staining (DeadEnd Fluorometric TUNEL System;
Promega Corporation, Madison, WI, USA). In brief, 4% formaldehyde
was used to fix cells at 4°C for 25 min. After the equilibration
for 10 min at room temperature, the cells were incubated with TdT
reaction mix at 37°C for 1 h. Afterwards, 2X SSC was added into
cells for 15 min, and then stained with DAPI solution for 5 min to
visualize all nuclei.
qPCR analysis
After various treatments, total RNAs was extracted
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) and
complementary DNA synthesis was carried out using miRNA specific
primers (Invitrogen; Thermo Fisher Scientific, Inc.). The PCR
primers for miR-4262 and U6 were commercially obtained from Applied
Biosystems (Foster City, CA, USA). The primers for uncoordinated
51-like kinase 1 (ULK1), asparagine-linked glycosylation 5 (ALG5),
Beclin-1, LC3II, type II collagen (COL2A1), aggrecan (ACAN), matrix
metallo protease 13 (MMP-13), a disintegrin and metalloproteinase
with thrombospondin motifs-5 (ADAMTS-5), SIRT1, and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are shown in
Table I. The PCR parameters were set
as follows: 95°C for 10 min, 40 cycles of 94°C for 30 sec, 58°C for
30 sec, and 72°C for 15 sec. U6 or GAPDH was used as the reference
genes, and relative gene expression level was calculated using
comparative Cquantification cycle method
(2−ΔΔCq).
 | Table I.Primer sequences for specific
genes. |
Table I.
Primer sequences for specific
genes.
| Gene | Primer
sequence |
|---|
| ULK | Forward:
5′-GTGCAGTCGGCTGCCCTGGAC-3′ |
|
| Reverse:
5′-TCAGGCACAGATGCCAGTCAGC-3′ |
| ALG5 | Forward:
5′-CATATGATGACTCGCACCCGCAAGCGCAC-3′ |
|
| Reverse: 5′-
AAGCTTCAGGAGTGTGTGACCTGCAGCTTG-3′ |
| Beclin-1 | Forward: 5′-
GAGGGATGGAAGGGTCTAAG-3′ |
|
| Reverse:
5′-GCCTGGGCTGTGGTAAGT-3′ |
| LC3II | Forward:
5′-GATGTCCGACTTATTCGAGAGC-3′ |
|
| Reverse:
5′-TTGAGCTGTAAGCGCCTTCTA-3′ |
| COL2A1 | Forward:
5′-GGCAATAGCAGGTTCACGTACA-3′ |
|
| Reverse:
5′-GATAACAGTCTTGCCCCACTTACC-3′ |
| ACAN | Forward:
5′-AGTCCTCAAGCCTCCTGTACTCA-3′ |
|
| Reverse:
5′-CGGGAAGTGGCGGTAACA-3′ |
| MMP-13 | Forward:
5′-CTTCTGGTCTTCTGGCACACG-3′ |
|
| Reverse:
5′-CCCCACCCCATACATCTGAAA-3′ |
| ADAMTS-5 | Forward:
5′-GGCGCAAATCCGGGTC-3′ |
|
| Reverse:
5′-CGCCATTCACGGTGCC-3′ |
| SIRT1 | Forward:
5′-CAACTTGTACGACGAAGAC-3′ |
|
| Reverse:
5′-TCATCACCGAACAGAAGG-3′ |
| GAPDH | Forward:
5′-GAAGGTGAAGGTCGGAGTC-3′ |
|
| Reverse:
5′-GAAGATGGTGATGGGATTTC-3′ |
Western blotting
Protein from cells was extracted using RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China) and
concentration was measured using the BCA Protein Quantitative Assay
(Beyotime Institute of Biotechnology). Total 50 µg protein sample
(per lane) was separated on SDS-PAGE gel, blotted onto PVDF
membranes, and blocked in 5% non-fat milk for 1 h. The membranes
were probed with mouse polyclonal antibodies to ULK1 (120 kDa;
1;1,000; ab128859; Abcam, Cambridge, UK), ALG5 (32 kDa; 1;1,000;
ab108327; Abcam), Beclin-1 (52 kDa; 1;1,000; ab62557; Abcam),
LC3II/LC3I (LC3-II, 17 kDa, LC3-II 19 kDa; 1;1,000; ab48394;
Abcam), COL2A1 (142 kDa; 1;1,000; ab34712; Abcam), ACAN (250 kDa;
1;1,000; ab36861; Abcam), MMP-13 (54 kDa; 1;1,000; ab39012; Abcam),
ADAMTS-5 (73 kDa; 1;1,000; ab41037; Abcam), SIRT1 (120 kDa;
1;1,000; ab110304; Abcam), phospho-PI3K (p-PI3K) (84 kDa; 1;1,000;
ab182651; Abcam), PI3K (123 kDa; 1;1,000; ab151549; Abcam), p-AKT
(65 kDa; 1;1,000; SAB4301414; Sigma-Aldrich; Merck KGaA), AKT (55
kDa; 1;1,000; SAB4500797; Sigma-Aldrich; Merck KGaA), p-mTOR (250
kDa; 1;1,000; ab137133; Abcam), mTOR (252 kDa; 1;2,000; ab2732;
Abcam), and GAPDH (36 kDa; 1;2,000; ab8245; Abcam) overnight at
4°C, respectively. After washed for three times with PBS, the
membranes incubated with appropriate second antibody IgG (H+L)-HRP
(125 kDa; 1:5,000; ab97051; Abcam) for 2 h at room temperature.
Ultimately, the proteins were detected with Enhanced
chemiluminescence (EMD Millipore, Billerica, MA, USA).
Densitometric analysis for the protein bands was performed by Image
J 1.48v (http://imagej.nih.gov/ij/, US
National Institutes of Health, Bethesda, MD, USA).
Target prediction and luciferase
reporter assay
The potential target gene of miR-4262 was predicted
by TargetScanHuman 7.1 (http://www.targetscan.org/vert_71/). The wildtype (WT)
3′UTR fragment of SIRT1 that can bind to miR-4262 or mutant (MUT)
3′UTR fragment was amplified from the genomic DNA and cloned into
the pGL3 vector. The chondrocytes were then co-transfected with
miR-4262 and luciferase reporter comprising WT or MUT 3′UTR of
SIRT1 for 48 h using Lipofectamine® 2000. The Dual
Luciferase Assay kit (Promega Corporation,) was used to measure the
activity of luciferase.
Statistical analysis
All experiments were performed in triplicate.
Statistical analysis was carried out using statistical analysis
software (SPSS 19.0; SPSS, Inc., Chicago, IL, USA). Data were
expressed as the mean ± standard deviation and analyzed by one-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
Primary chondrocytes were successfully
isolated
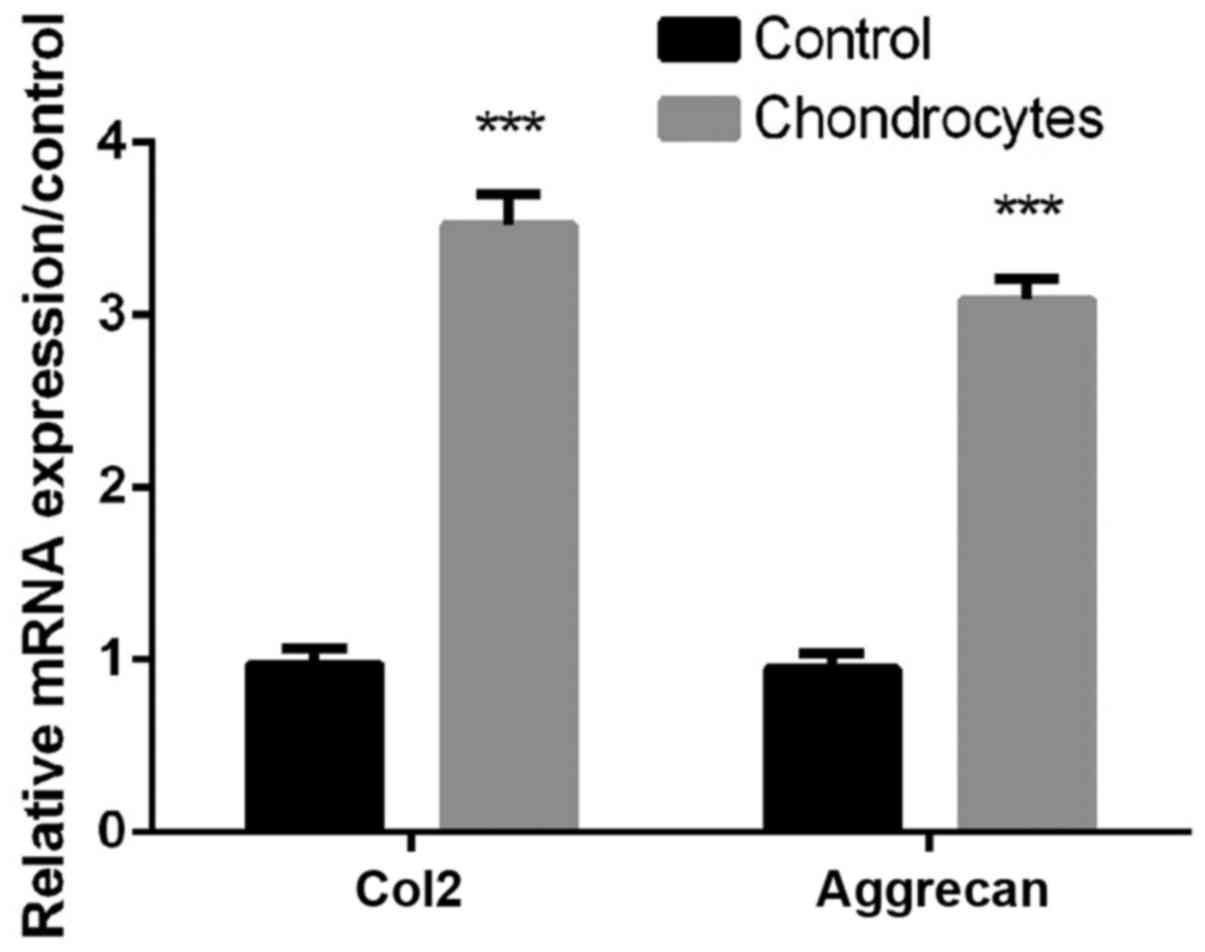
As shown in Fig. 1,
the expression levels of Col2 and aggrecan in chondrocytes were
significantly higher than controls (all P<0.01, Fig. 1), indicating that Primary
chondrocytes were successfully isolated for subsequent
treatments.
TNF-α decreases cell viability and
autophagy, and increases apoptosis and miR-4262 level in
chondrocytes
Compared with control, cell viability was
significantly inhibited, while the ratio of cell apoptosis was
markedly increased after treatment with TNF-α for 12, 24, and 48 h
(all P<0.05, Fig. 2A and B).
Compared with control group, the expression levels of
autophagy-related proteins, including ULK1, ALG5, Beclin-1, and
LC3II/LC3I, were remarkably lower in TNF-αgroup, while
significantly increased in RAPA group (all P<0.05, Fig. 2C). Nevertheless, only the expression
ratio of LC3II/LC3I was significantly decreased in TNF-α-treated
cells after treatment with lysosomal protease inhibitors E64d and
pepstatin A (P<0.05, Fig. 2C),
but not the expression levels of ULK1, ALG5, and Beclin-1. In
addition, TNF-α treatment significantly increased miR-4262 level
compared with control (all P<0.05, Fig. 2D).
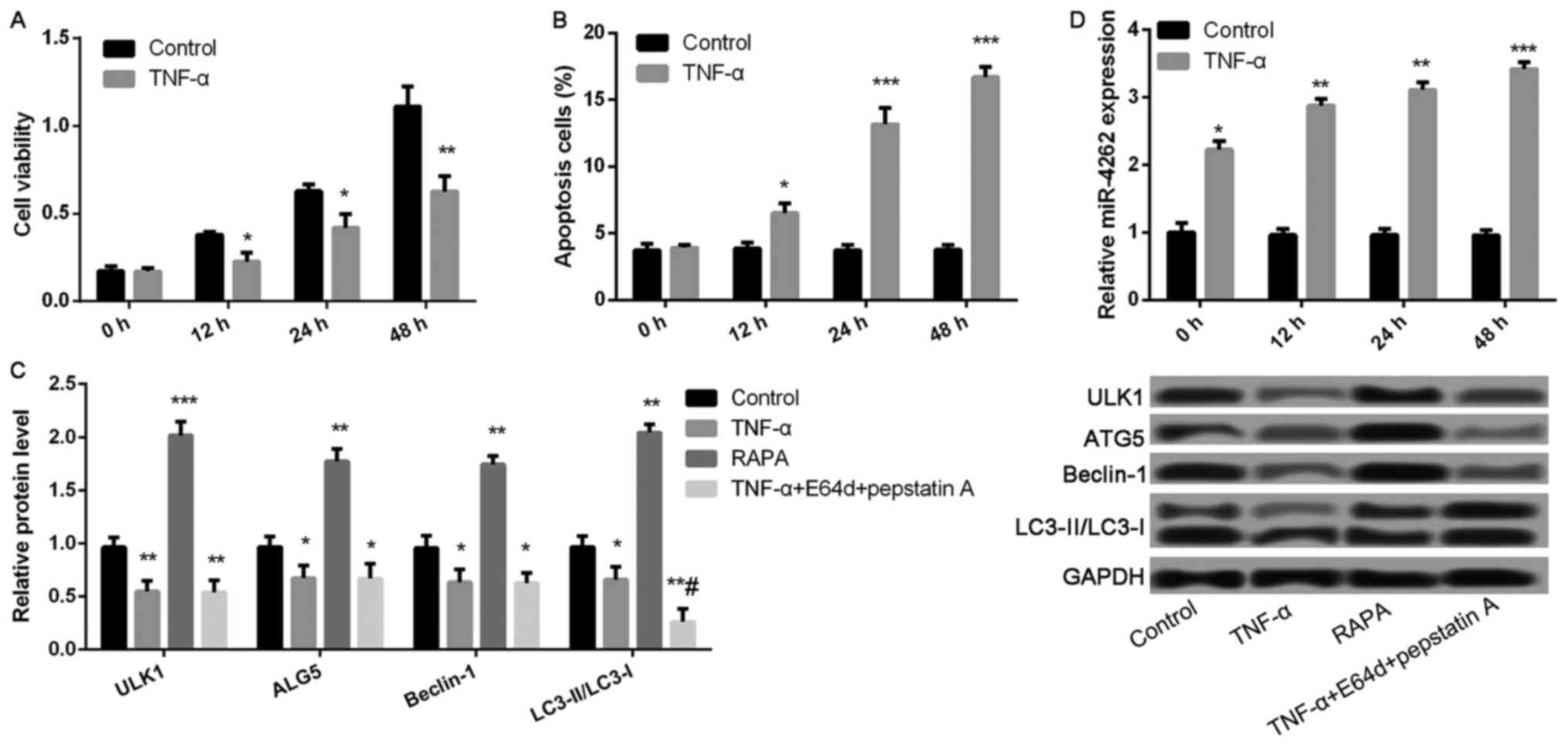 | Figure 2.TNF-α treatment significantly
decreases cell viability and autophagy, as well as increases
apoptosis and the expression of miR-4262 in chondrocytes. (A) The
cell viability in control and TNF-α groups at 0, 12, 24, 48 h using
CCK-8; (B) The cell apoptosis in control and TNF-α groups at 0, 12,
24, 48 h using TUNEL staining; (C) The expression of
autophagy-related proteins, including ULK1, ALG5, Beclin-1,
LC3II/LC3I, in control, TNF-α, RAPA and TNF-α + E64d + pepstatin A
groups using western blotting; (D) The level of miR-4262 in control
and TNF-α groups at 0, 12, 24, 48 h using quantitative reverse
transcription PCR. Three independent experiments were performed in
each assay. *P<0.05, **P<0.01, and ***P<0.001 compared
with the control group, #P<0.05 compared with the
TNF-α group. TNF-α, tumor necrosis factor-α; RAPA, rapamycin; ULK1,
uncoordinated 51-like kinase 1; ALG5, asparagine-linked
glycosylation 5. |
Overexpression of miR-4262 inhibits
cell viability, autophagy, and matrix synthesis, and promoted cell
apoptosis in chondrocytes
As shown in Fig. 3A,
the miR-4262 level was significantly increased in cells with
miR-4262 mimic compared with cells with mimic control (P<0.001),
and the transfection of miR-4262 inhibitor obviously inhibited the
miR-4262 level compared with the transfection of inhibitor control
(P<0.01). Moreover, compared with mimic control, cell viability
was significantly inhibited in miR-4262 mimic group, while the
percentage of apoptotic cells was obviously increased (all
P<0.05, Fig. 3B and C). The
expression levels of autophagy-related proteins, including ULK1,
ALG5, Beclin-1, and LC3II/LC3I, were all significantly decreased in
miR-4262 mimic group compared with mimic control group (all
P<0.05, Fig. 3D). Opposite
effects on cell viability, apoptosis and autophagy were obtained
after transfection with miR-4262 inhibitor (all P<0.05, Fig. 3B-D). Furthermore, the results
revealed that, compared with TNF-α + mimic control group, cell
viability was significantly inhibited in cells with the treatments
of TNF-α and miR-4262, while the ratio of cell apoptosis was
markedly increased (all P<0.05, Fig.
3E and F). The expression levels of autophagy-related proteins,
including ULK1, ALG5, Beclin-1, and LC3II/LC3I, were all remarkably
lower expressed in cells with the treatments of TNF-α and miR-4262
(all P<0.01, Fig. 3G). Notably,
compared with TNF-α-treated cells, the expression ratio of
LC3II/LC3I was further decreased after treatment with lysosomal
protease inhibitors E64d and pepstatin A (P<0.05, Fig. 3G). In addition, we detect the
expression of matrix synthesis-related proteins, such as COL2A1,
ACAN, MMP-13 and ADAMTS-5. The results showed, compared with TNF-α
+ mimic control group, that the protein levels of COL2A1 and ACAN
were significantly reduced in cells with the treatments of TNF-α
and miR-4262, while MMP-13 and ADAMTS-5 levels were remarkably
increased (all P<0.01, Fig. 3H).
Consistently, cells with the treatments of TNF-α and miR-4262
inhibitor exhibited the opposite effects on cell viability, cell
apoptosis, cell autophagy, and matrix synthesis in chondrocytes
(all P<0.05, Fig. 3E-H).
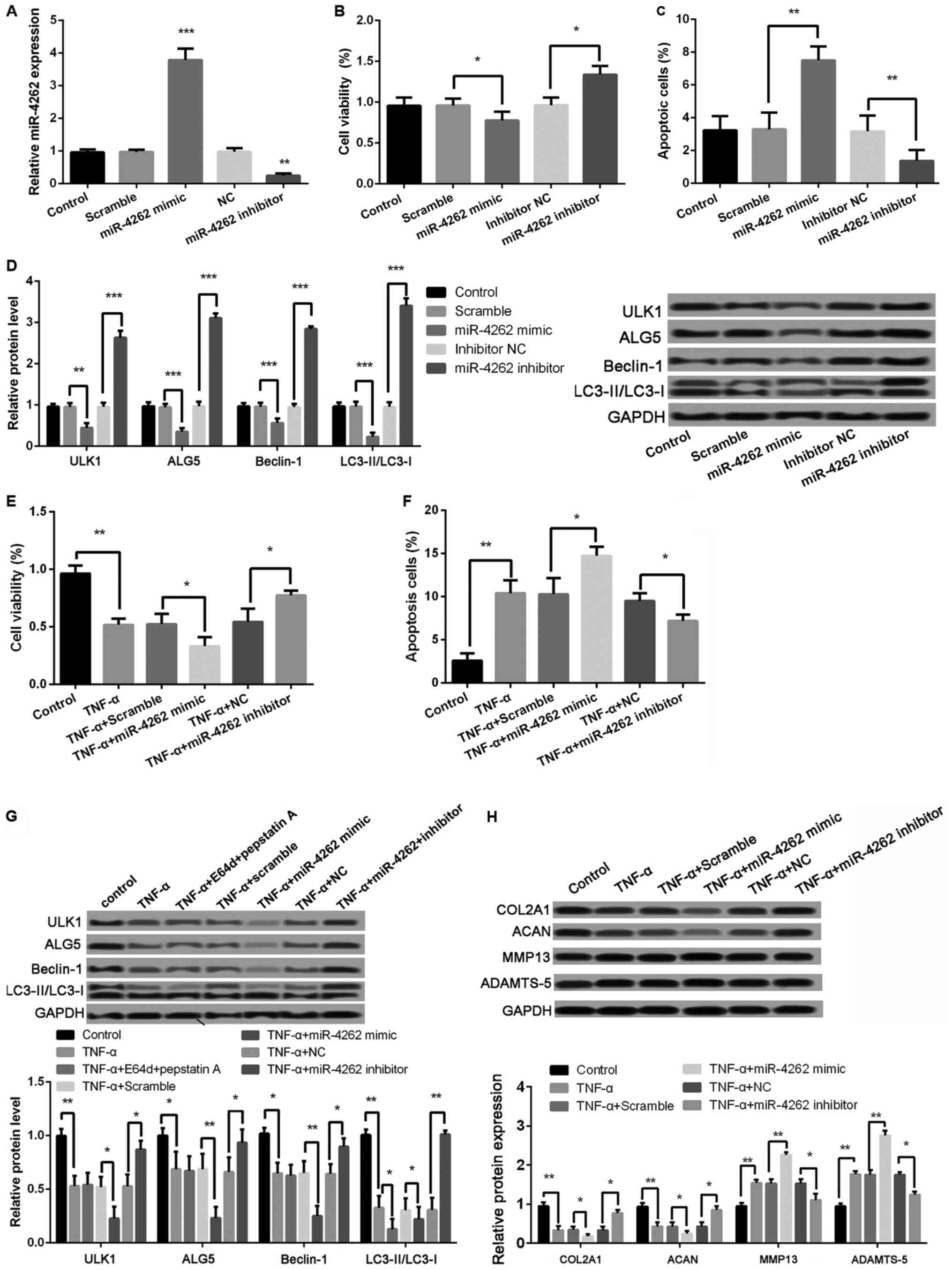 | Figure 3.Upregulated miR-4262 further
decreases cell viability, autophagy, and matrix synthesis as well
as increases apoptosis in TNF-α-treated chondrocytes. (A) The
miR-4262 level in control, scramble (control of mimic), miR-4262
mimic, NC (control of inhibitor), and miR-4262 inhibitor groups by
qPCR; (B) The cell viability in control, scramble, miR-4262 mimic,
inhibitor NC, and miR-4262 inhibitor groups using CCK-8; (C) The
cell apoptosis in control, scramble, miR-4262 mimic, inhibitor NC,
and miR-4262 inhibitor groups using TUNEL staining; (D) The
expression of autophagy-related proteins, including ULK1, ALG5,
Beclin-1, LC3II/LC3I, in control, scramble, miR-4262 mimic,
inhibitor NC, and miR-4262 inhibitor groups using western blotting;
(E) The cell viability in control, TNF-α, TNF-α + scramble, TNF-α +
miR-4262 mimic, TNF-α + NC, and TNF-α + miR-4262 inhibitor groups
using CCK-8; (F) The cell apoptosis in control, TNF-α, TNF-α +
scramble, TNF-α + miR-4262 mimic, TNF-α + NC, and TNF-α + miR-4262
inhibitor groups using TUNEL staining; (G) The expression of
autophagy-related proteins, including ULK1, ALG5, Beclin-1,
LC3II/LC3I, in control, TNF-α, TNF-α+E64d+pepstatin (A) TNF-α +
scramble, TNF-α + miR-4262 mimic, TNF-α + NC, and TNF-α + miR-4262
inhibitor groups using western blotting; (H) The expression of
matrix synthesis-related proteins, such as COL2A1, ACAN, MMP-13 and
ADAMTS-5, in control, TNF-α, TNF-α + scramble, TNF-α + miR-4262
mimic, TNF-α + NC, and TNF-α + miR-4262 inhibitor groups using
western blotting. Three independent experiments were performed in
each assay. *P<0.05, **P<0.01, and ***P<0.001 TNF-α, tumor
necrosis factor-α; ULK1, uncoordinated 51-like kinase 1; ALG5,
asparagine-linked glycosylation 5; COL2A1, type II collagen; ACAN,
aggrecan; MMP-13, matrix metallo protease 13; ADAMTS-5, a
disintegrin and metalloproteinase with thrombospondin motifs-5. |
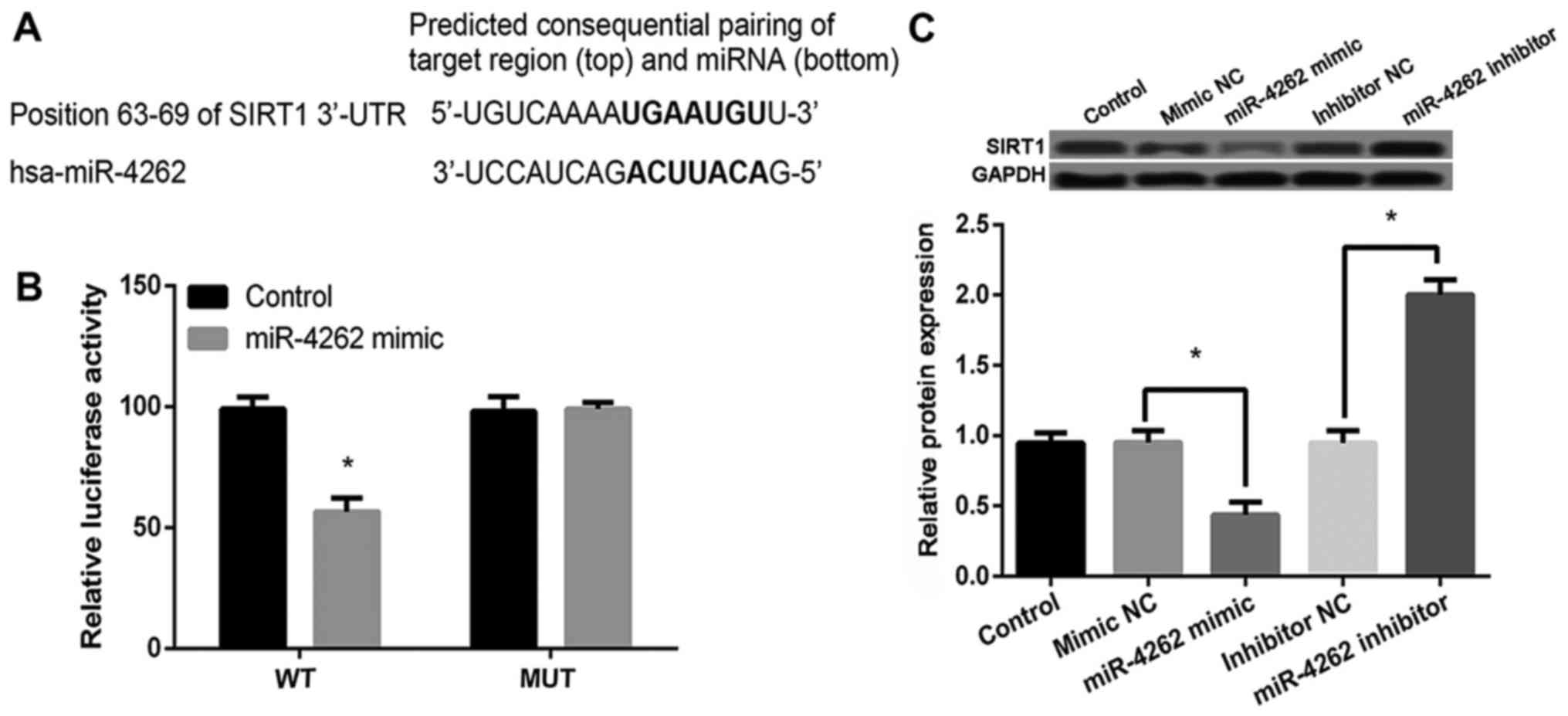
SIRT1 is confirmed as a direct target
of miR-4262
Sequence analysis with TargetScanHuman revealed that
SIRT1 was a potential target gene of miR-4262 (http://www.targetscan.org/cgi-bin/targetscan/vert_71/view_gene.cgi?rs=ENST00000212015.6&taxid=9606&members=miR-181-5p&showcnc=0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1;
Fig. 4A). Then luciferase reporter
assay showed that miR-4262 mimic significantly inhibited the
luciferase activity of WT 3′UTR of SIRT1 (P<0.05) but not MUT
3′UTR of SIRT1 compared with control (Fig. 4B). In addition, compared with cells
with mimic or inhibitor control, miR-4262 mimic or inhibitor
obviously inhibited or increased the expression of SIRT1
(P<0.05, Fig. 4C). These data
indicated that SIRT1 was a target of miR-4262 and its expression
was negatively regulated by miR-4262.
Overexpression of SIRT1 promotes cell
viability, autophagy, and matrix synthesis, and inhibited cell
apoptosis, autophagy, and matrix synthesis in chondrocytes
As shown in Fig. 5A and
B, the protein and mRNA levels of SIRT1 were significantly
increased in cells with pEX-SIRT1 compared with cells with pEX
(P<0.001), and the transfection of sh-SIRT1 obviously inhibited
the SIRT1 level compared with the transfection of siNC (P<0.05).
In addition, compared with TNF-α + pEX treated cells, cell
viability was significantly increased, while the ratio of cell
apoptosis was obviously inhibited in cells with the treatments of
TNF-α and pEX-SIRT1 (all P<0.05, Fig.
5C and D). The expression levels of autophagy-related proteins,
including ULK1, ALG5, Beclin-1, and LC3II/LC3I, were all remarkably
increased in cells with the treatments of TNF-α and pEX-SIRT1 (all
P<0.01, Fig. 5E) compared with
TNF-α + pEX treated cells. In addition, the protein levels of
COL2A1 and ACAN were significantly increased, while MMP-13 and
ADAMTS-5 levels were remarkably reduced in cells with the
treatments of TNF-α and pEX-SIRT1 (all P<0.01, Fig. 5F) compared with TNF-α + pEX treated
cells. Consistently, cells with the treatments of TNF-α and
miR-4262 inhibitor exhibited the opposite effects on cell
viability, cell apoptosis, cell autophagy, and matrix synthesis in
chondrocytes (all P<0.05, Fig.
3E-H). Consistently, cells with the treatments of TNF-α and
sh-SIRT1 exhibited the opposite effects on cell viability, cell
apoptosis, cell autophagy, and matrix synthesis in chondrocytes
(all P<0.05, Fig. 5C and D).
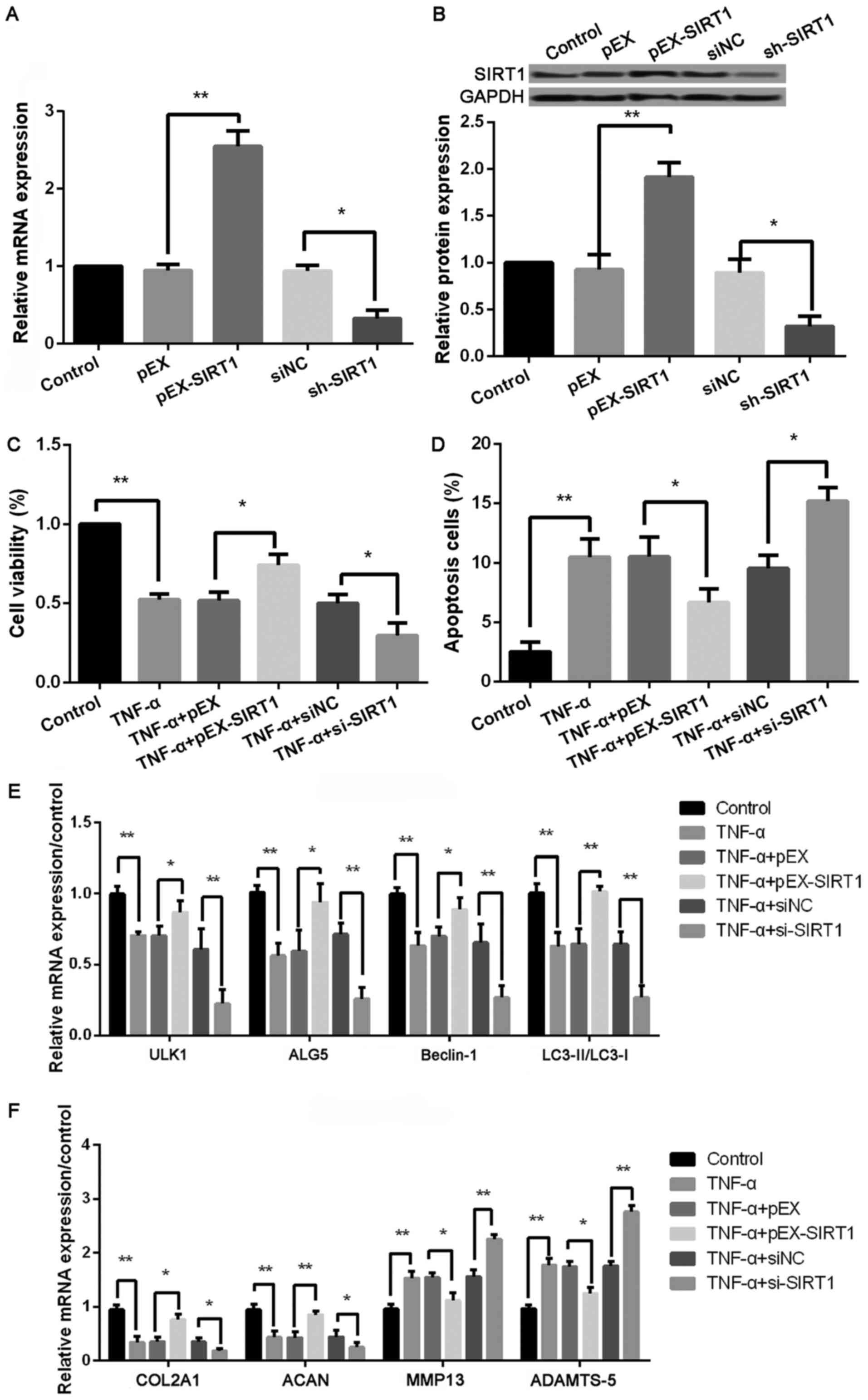 | Figure 5.Upregulated SIRT1 increases cell
viability, autophagy, and matrix synthesis as well as decreases
apoptosis in TNF-α-treated chondrocytes. (A) The SIRT1 level in
control, pEX, pEX-SIRT1, siNC, and si-SIRT1 groups by qPCR; (B) The
SIRT1 level in control, pEX, pEX-SIRT1, siNC, and si-SIRT1 groups
by western blotting; The cell viability in (C) control, TNF-α,
TNF-α + pEX, TNF-α + pEX-SIRT1, TNF-α + siNC, and TNF-α + si-SIRT1
groups using CCK-8; (D) The cell apoptosis in control, TNF-α, TNF-α
+ pEX, TNF-α + pEX-SIRT1, TNF-α + siNC, and TNF-α + si-SIRT1 groups
using TUNEL staining; (E) The expression of autophagy-related
proteins, including ULK1, ALG5, Beclin-1, LC3II/LC3I, in control,
TNF-α, TNF-α + pEX, TNF-α + pEX-SIRT1, TNF-α + siNC, and TNF-α +
si-SIRT1 groups using qPCR; (F) The expression of matrix
synthesis-related proteins, such as COL2A1, ACAN, MMP-13 and
ADAMTS-5, in control, TNF-α, TNF-α + pEX, TNF-α + pEX-SIRT1, TNF-α
+ siNC, and TNF-α + si-SIRT1 groups using qPCR. Three independent
experiments were performed in each assay. *P<0.05, and
**P<0.01 TNF-α, tumor necrosis factor-α; ULK1, uncoordinated
51-like kinase 1; ALG5, asparagine-linked glycosylation 5; COL2A1,
type II collagen; ACAN, aggrecan; MMP-13, matrix metallo protease
13; ADAMTS-5, a disintegrin and metalloproteinase with
thrombospondin motifs-5; SIRT1, sirtuin type 1. |
Roles of miR-4262 in chondrocytes are
associated with the regulation of SIRT1
The results revealed that compared with cells with
TNF-α, miR-4262 mimic and pEX, cell viability was significantly
increased, while the ratio of cell apoptosis was obviously
inhibited in cells with the treatments of TNF-α, miR-4262 mimic and
pEX-SIRT1 (all P<0.05, Fig. 6A and
B). The expression levels of autophagy-related proteins,
including ULK1, ALG5, Beclin-1, and LC3II/LC3I, were all remarkably
higher expressed in cells with the co-treatments of TNF-α, miR-4262
mimic and pEX-SIRT1 than those in cells with TNF-α, miR-4262 mimic
and pEX (all P<0.05, Fig. 6C). In
addition, protein levels of COL2A1 and ACAN were significantly
increased, while MMP-13 and ADAMTS-5 levels were remarkably reduced
in cells with the co-treatments of TNF-α, miR-4262 mimic and
pEX-SIRT1 compared with cells with TNF-α, miR-4262 mimic and pEX
(all P<0.01, Fig. 6D).
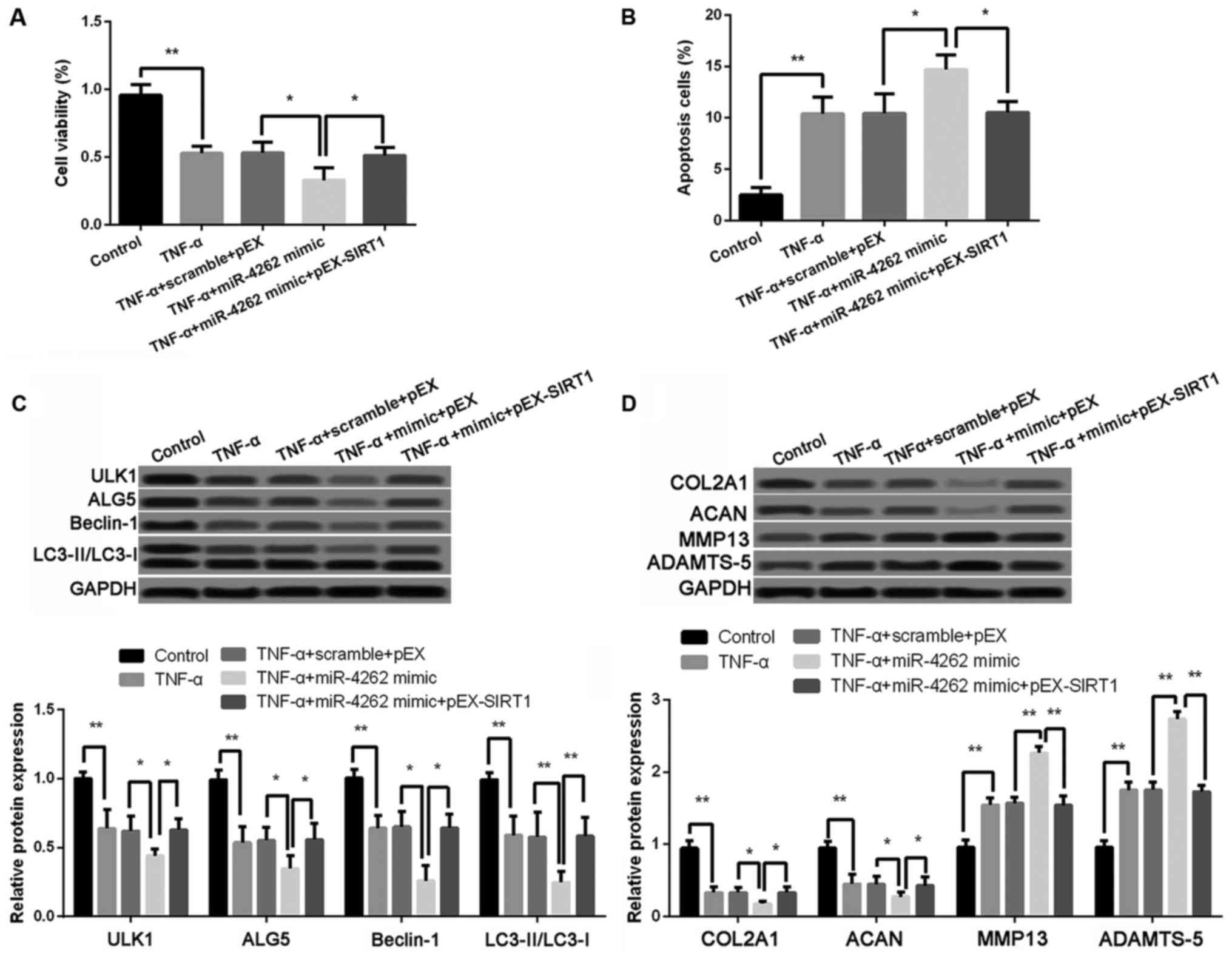 | Figure 6.Effects of miR-4262 on cell
viability, cell apoptosis, cell autophagy, and matrix synthesis is
inhibited by SIRT1. (A) The cell viability in control, TNF-α, TNF-α
+ scramble + pEX, TNF-α + miR-4262 mimic, and TNF-α + miR-4262
mimic + pEX-SIRT1 groups using CCK-8; (B) The cell apoptosis in
control, TNF-α, TNF-α + scramble + pEX, TNF-α + miR-4262 mimic, and
TNF-α + miR-4262 mimic + pEX-SIRT1 groups using TUNEL staining; (C)
The expression of autophagy-related proteins, including ULK1, ALG5,
Beclin-1, LC3II/LC3I, in control, TNF-α, TNF-α + scramble + pEX,
TNF-α + miR-4262 mimic, and TNF-α + miR-4262 mimic + pEX-SIRT1
groups using western blotting; (D) The expression of matrix
synthesis-related proteins, such as COL2A1, ACAN, MMP-13 and
ADAMTS-5, in control, TNF-α, TNF-α + scramble + pEX, TNF-α +
miR-4262 mimic, and TNF-α + miR-4262 mimic + pEX-SIRT1 groups using
western blotting. Three independent experiments were performed in
each assay. *P<0.05, and **P<0.01 TNF-α, tumor necrosis
factor-α; ULK1, uncoordinated 51-like kinase 1; ALG5,
asparagine-linked glycosylation 5; COL2A1, type II collagen; ACAN,
aggrecan; MMP-13, matrix metallo protease 13; ADAMTS-5, a
disintegrin and metalloproteinase with thrombospondin motifs-5;
SIRT1, sirtuin type 1. |
Overexpression of miR-4262 activates
PI3K/AKT/mTOR signaling pathway
Compared with untreated cells, the expression levels
of p-PI3K, p-AKT, and p-mTOR was significantly increased in TNF-α
treated cells, and the co-treatments of TNF-α and miR-4262 mimic
further increased the expression levels of p-PI3K, p-AKT, and
p-mTOR (all P<0.05, Fig. 7). In
addition, cells with TNF-α, miR-4262 mimic and pEX-SIRT1 exhibited
lower levels of p-PI3K, p-AKT, and p-mTOR than those in cells with
TNF-α, miR-4262 mimic and pEX (all P<0.05, Fig. 7).
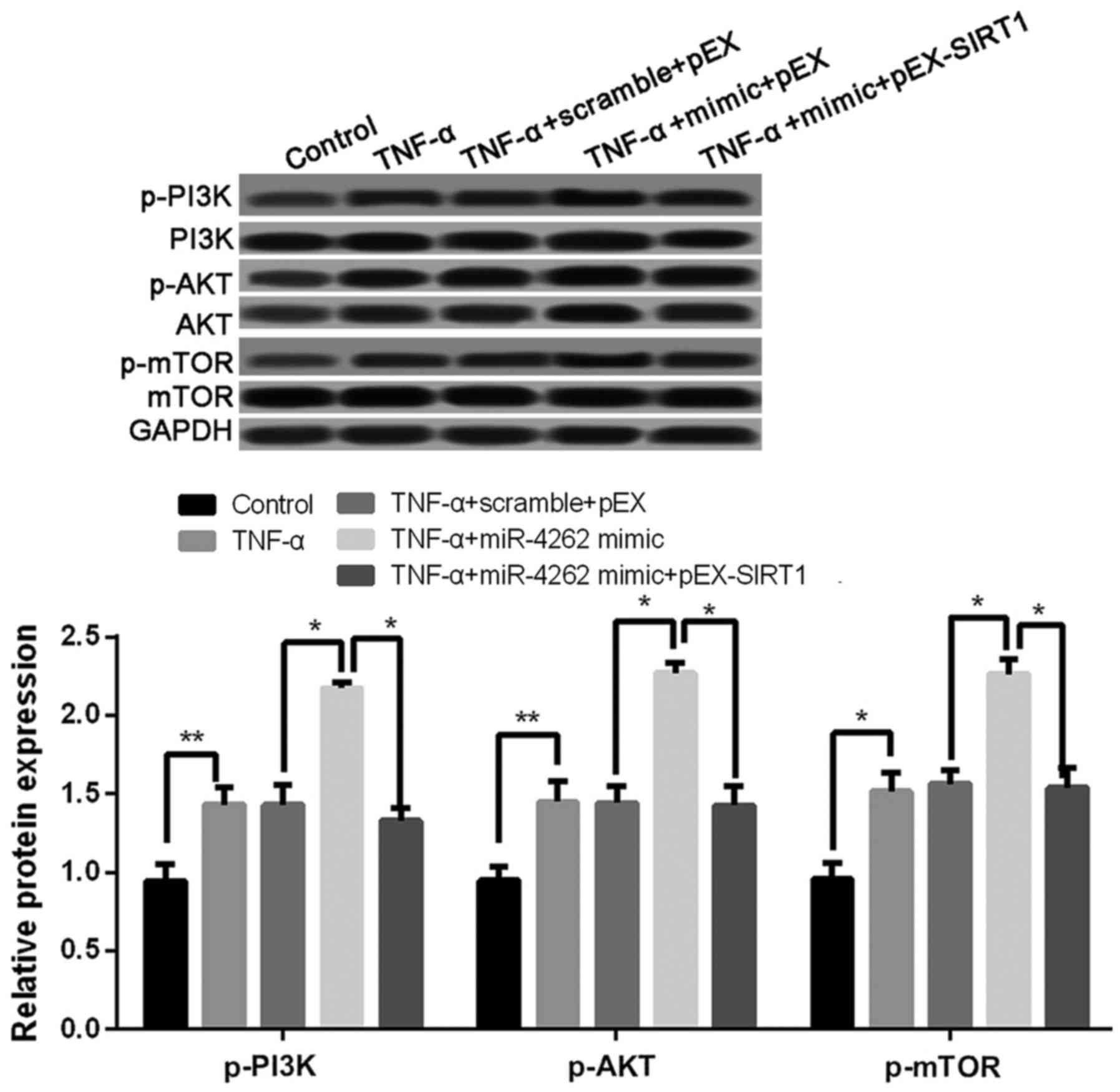 | Figure 7.Upregulated miR-4262 activates
PI3K/AKT/mTOR signaling pathway in TNF-α-treated chondrocytes. The
expression of p-PI3K, p-AKT, and p-mTOR in control, TNF-α, TNF-α +
scramble + pEX, TNF-α + miR-4262 mimic, and TNF-α + miR-4262 mimic
+ pEX-SIRT1 groups using western blotting. Three independent
experiments were performed in each assay. *P<0.05, and
**P<0.01 TNF-α, tumor necrosis factor-α; phosphoinositide
3-kinase; phospho-PI3K (p-PI3K), PI3K; mTOR, mammalian target of
rapamycin; SIRT1, sirtuin type 1. |
Discussion
In the current study, our results showed that TNF-α
could inhibit cell viability and the expression levels of
autophagy-related proteins as well as increase cell apoptosis in
rat chondrocytes. Overexpressed miR-4262 further promoted the cell
injury induced by TNF-α, while its target gene SIRT1 could
alleviate the cell injury by promoting cell viability and the
expression levels of autophagy-related proteins, as well as
inhibiting cell apoptosis in TNF-α-treated chondrocytes. In
addition, miR-4262 influenced the expression levels of matrix
synthesis-related proteins. Furthermore, up-regulated miR-4262
remarkably increased the expression of p-PI3K, p-AKT and
p-mTOR.
Several studies had demonstrated that miRNAs played
a vital role in the regulation of the OA development, including
miR-210 (21), miR-16 (22), miR-21 (23), and miR-142 (24). This study found the up-regulation of
miR-4262 in TNF-α-treated chondrocytes. Few studies had
investigated the roles of miR-4262 in OA. Previous studies had
revealed the proliferation promotion effect of miR-4262
overexpression in cancers, including melanoma (14), breast cancer (25), hepatocellular carcinoma (26), and osteosarcoma (16), indicating an carcinogenesis role of
miR-4262. However, this study found that up-regulated miR-4262
inhibited cell viability in TNF-α-treated chondrocytes, which might
be explained that miR-4262 exerted the opposite effect on cell
proliferation in cancers and non-cancers.
It was reported that OA is caused by the death of
chondrocytes and the loss of extracellular matrix in cartilage
degeneration (27). Apoptosis and
autophagy are basic physiologic processes for balancing cell
homeostasis and death. Apoptosis of damaged chondrocytes led to
articular cartilage degeneration and subsequent OA development,
while autophagy could inhibit damaged chondrocyte apoptosis and
thus alleviate the progression of OA (28). In this study, up-regulated miR-4262
inhibited the expression levels of autophagy-related proteins and
increased cell apoptosis in TNF-α-treated chondrocytes. It was
well-known that TNF-α played a vital role in the pathogenesis of
OA, and participated in the progression of cartilage degeneration
(29). TNF-α could induce articular
chondrocytes death through promoting cell apoptosis and autophagy
(30–32). Several studies also found that cell
autophagy had a protective effect on chondrocytes by inhibiting
cell death (33,34). Huang et al (35) demonstrated that leptin was implicated
in OA pathogenesis though promoting apoptosis and inhibiting
autophagy of chondrocytes. This study showed that TNF-α inhibited
cell autophagy and increased cell apoptosis, and up-regulated
miR-4262 enhanced these roles in TNF-α-treated chondrocytes,
implying that up-regulation of miR-4262 might contribute to OA
development in rats via promoting apoptosis and inhibiting
autophagy of chondrocytes. Meanwhile, this study revealed that both
miR-4262 overexpression and TNF-α treatment inhibited the levels of
COL2A1 and ACAN, while increased MMP-13 and ADAMTS-5 levels.
Consistent with this study, a recent study further demonstrated
that TNF-α also had been proved to be associated with the loss of
matrix synthesis-related proteins, such as COL2A1 and ACAN, as well
as the elevation of MMP-13 and ADAMTS-5 (36). All these results indicated that
miR-4262 might be involved in the development of OA in rats through
regulating cell viability, cell apoptosis, cell autophagy, and
matrix synthesis in chondrocytes.
Furthermore, this study confirmed the target gene of
miR-4262 was SIRT1. SIRT1 is found to exert deacetylation for
target substrates, and then participate in cell survival,
metabolism, and oxygen consumption. Recently, SIRT1 was proved to
be down-regulated in chondrocytes from OA (37), which was consistent with our study.
Previous study suggested that SIRT1 could inhibit the apoptosis of
chondrocytes and increase the survival of OA (38). SIRT1 also was reported to inhibit the
extracellular matrix degradation by inhibiting MMP13 production
(20). Similarly, this study
revealed that SIRT1 inhibited cell apoptosis and increased the
extracellular matrix in TNF-α-treated chondrocytes. Meanwhile,
SIRT1 overexpression could reverse the effects of miR-4262 on
TNF-α-treated chondrocytes. The above results promoted that the
role of miR-4262 in TNF-α-treated chondrocytes might be regulated
by its target gene SIRT1.
To further investigate the mechanism of miR-4262 in
OA, we focused on the PI3K/AKT/mTOR signaling pathway.
PI3K/AKT/mTOR signaling pathway was not only involved in the
regulation of inflammatory processes (39), but also regulated many normal
cellular processes, such as cell proliferation and apoptosis
(40). The activations of PI3K could
induce the phosphorylation of AKT, and then inhibited the
expressions of Bcl-2 and Bcl-xL (anti-apoptotic proteins), as well
as Bax and Bak (pro-apoptotic proteins) (40). In addition, PI3K/Akt pathway could
increase MMP levels produced by chondrocytes, while inhibition of
mTOR increased cell autophagy, and then inhibit chondrocyte death
(41). The present study found that
that up-regulated miR-4262 increased the expressions of p-PI3K,
p-AKT, and p-mTOR in TNF-α-treated chondrocytes, while it could be
reversed by SIRT1. Therefore, we speculated that the role of
miR-4262 overexpression in TNF-α-treated chondrocytes might be
mediated by the activation of PI3K/AKT/mTOR signaling pathway.
In conclusion, the present study reveals that
miR-4262 may promote the occurrence and development of OA in rats
by regulating cell viability, cell apoptosis, cell autophagy, and
matrix synthesis. Furthermore, these roles of miR-4262 may be
associated with PI3K/AKT/mTOR signaling pathway.
References
|
1
|
Glynjones S, Palmer AJ, Agricola R, Price
AJ, Vincent TL, Weinans H and Carr AJ: Osteoarthritis. Lancet.
386:376–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neogi T: The epidemiology and impact of
pain in osteoarthritis. Osteoarthritis Cartilage. 21:1145–1153.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wilkins JM, Loughlin J and Snelling SJ:
Osteoarthritis genetics: Current status and future prospects.
Future Rheumatol. 2:607–620. 2015. View Article : Google Scholar
|
|
4
|
Schachar R and Ogilvie-Harris D:
Osteoarthritis: Joint conservation strategiesOsteoarthritis. Kapoor
M and Mahomed N: Springer International Publishing; Switzerland:
2015, View Article : Google Scholar
|
|
5
|
Litwic A, Edwards MH, Dennison EM and
Cooper C: Epidemiology and burden of osteoarthritis. Br Med Bull.
105:185–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jovanovic M and Hengartner M: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saito Y, Saito H, Liang G and Friedman JM:
Epigenetic alterations and MicroRNA misexpression in cancer and
autoimmune diseases: A critical review. Clin Rev Allergy Immunol.
47:128–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Condorelli G, Latronico MV and Cavarretta
E: microRNAs in cardiovascular diseases: Current knowledge and the
road ahead. J Am Coll Cardiol. 63:2177–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan L, Yu JT and Tan L: Causes and
consequences of MicroRNA dysregulation in neurodegenerative
diseases. Mol Neurobiol. 51:1249–1262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji Y, Gao F, Sun B, Hao J and Liu Z:
Angiotensin-converting enzyme 2 inhibits apoptosis of pulmonary
endothelial cells during acute lung injury through suppressing
SMAD2 phosphorylation. Cell Physiol Biochem. 35:2203–2212. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang D, Li Z, Zhang Y, Tu C, Huo J and
Liu Y: miR-4262 promotes the proliferation of human cutaneous
malignant melanoma cells through KLF6-mediated EGFR inactivation
and p21 upregulation. Oncol Rep. 36:3657–3663. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu S, Wu J, Gao Y, Han G, Ding W and Huang
X: MicroRNA-4262 activates the NF-κB and enhances the proliferation
of hepatocellular carcinoma cells. Int J Biol Macromol. 86:43–49.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song K, Liu N, Yang Y and Qiu X:
Regulation of osteosarcoma cell invasion through osteopontin
modification by miR-4262. Tumour Biol. 37:6493–6499. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fujita N, Matsushita T, Ishida K, Kubo S,
Matsumoto T, Takayama K, Kurosaka M and Kuroda R: Potential
involvement of SIRT1 in the pathogenesis of osteoarthritis through
the modulation of chondrocyte gene expressions. J Orthop Res.
29:511–515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takayama K, Ishida K, Matsushita T, Fujita
N, Hayashi S, Sasaki K, Tei K, Kubo S, Matsumoto T, Fujioka H, et
al: SIRT1 regulation of apoptosis of human chondrocytes. Arthritis
Rheum. 60:2731–2740. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsuzaki T, Matsushita T, Takayama K,
Matsumoto T, Nishida K, Kuroda R and Kurosaka M: Disruption of
Sirt1 in chondrocytes causes accelerated progression of
osteoarthritis under mechanical stress and during ageing in mice.
Ann Rheum Dis. 73:1397–1404. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moon MH, Jeong JK, Lee YJ, Seol JW,
Jackson CJ and Park SY: SIRT1, a class III histone deacetylase,
regulates TNF-α-induced inflammation in human chondrocytes.
Osteoarthritis Cartilage. 21:470–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang D, Cao X, Li J and Zhao G: MiR-210
inhibits NF-κB signaling pathway by targeting DR6 in
osteoarthritis. Sci Rep. 5:127752015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Jia J, Liu X, Yang S, Ye S, Yang W
and Zhang Y: MicroRNA-16-5p controls development of osteoarthritis
by targeting SMAD3 in chondrocytes. Curr Pharm Des. 21:5160–5167.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Jie J, Yang S, Liu X, Ye S and
Tian H: MicroRNA-21 controls the development of osteoarthritis by
targeting GDF-5 in chondrocytes. Exp Mol Med. 46:e792014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Guo Y, Wang C and Yu H, Yu X and
Yu H: MicroRNA-142-3p inhibits chondrocyte apoptosis and
inflammation in osteoarthritis by targeting HMGB1. Inflammation.
39:1718–1728. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang K, Ren Y, Liu Y, Zhang J and He JJ:
miR-4262 promotes proliferation and invasion of human breast cancer
cells through directly targeting KLF6 and KLF15. Oncol Res.
25:277–283. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu W, Xiao L, Cao C, Hua S and Wu D: UBE2T
promotes nasopharyngeal carcinoma cell proliferation, invasion, and
metastasis by activating the AKT/GSK3β/β-catenin pathway.
Oncotarget. 7:15161–15172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Almonte-Becerril M, Navarro-Garcia F,
Gonzalez-Robles A, Vega-Lopez MA, Lavalle C and Kouri JB: Cell
death of chondrocytes is a combination between apoptosis and
autophagy during the pathogenesis of Osteoarthritis within an
experimental model. Apoptosis. 15:631–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu LG, Xu C and Yilihamu T: Autophagy
genes associated with chondrocyte apoptosis: protection and
balancing effects. Chin J Tissue Eng Res. 19:3231–3235. 2015.(In
Chinese).
|
|
29
|
Kapoor M, Martelpelletier J, Lajeunesse D,
Pelletier JP and Fahmi H: Role of proinflammatory cytokines in the
pathophysiology of osteoarthritis. Nat Rev Rheumatol. 7:33–42.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aizawa T, Kon T, Einhorn TA and
Gerstenfeld LC: Induction of apoptosis in chondrocytes by tumor
necrosis factor-alpha. J Orthop Res. 19:785–796. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stanic I, Facchini A, Borzì RM, Vitellozzi
R, Stefanelli C, Goldring MB, Guarnieri C, Facchini A and Flamigni
F: Polyamine depletion inhibits apoptosis following blocking of
survival pathways in human chondrocytes stimulated by tumor
necrosis factor-alpha. J Cell Physiol. 206:138–146. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee SW, Song YS, Lee SY, Yoon YG, Lee SH,
Park BS, Yun I, Choi H, Kim K, Chung WT and Yoo YH: Downregulation
of protein kinase CK2 activity facilitates tumor necrosis
factor-α-mediated chondrocyte death through apoptosis and
autophagy. PLoS One. 6:e191632011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bohensky J, Shapiro IM, Leshinsky S,
Watanabe H and Srinivas V: PIM-2 is an independent regulator of
chondrocyte survival and autophagy in the epiphyseal growth plate.
J Cell Physiol. 213:246–251. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bohensky J, Shapiro IM, Leshinsky S,
Terkhorn SP, Adams CS and Srinivas V: HIF-1 regulation of
chondrocyte apoptosis: Induction of the autophagic pathway.
Autophagy. 3:207–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang ZM, Du SH, Huang LG, Li JH, Xiao L
and Tong P: Leptin promotes apoptosis and inhibits autophagy of
chondrocytes through upregulating lysyl oxidase-like 3 during
osteoarthritis pathogenesis. Osteoarthritis Cartilage.
24:1246–1253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goldring MB: Molecular regulation of the
chondrocyte phenotype. J Musculoskelet Neuronal Interact.
2:517–520. 2002.PubMed/NCBI
|
|
37
|
Dvir-Ginzberg M, Gagarina V, Lee EJ and
Hall DJ: Regulation of cartilage-specific gene expression in human
chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J
Biol Chem. 283:36300–36310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gagarina V, Gabay O, Dvir-Ginzberg M, Lee
EJ, Brady JK, Quon MJ and Hall DJ: SirT1 enhances survival of human
osteoarthritic chondrocytes by repressing protein tyrosine
phosphatase 1B and activating the insulin-like growth factor
receptor pathway. Arthritis Rheum. 62:1383–1392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu CQ, Liu BJ, Wu JF, Xu YC, Duan XH, Cao
YX and Dong JC: Icariin attenuates LPS-induced acute inflammatory
responses: Involvement of PI3K/Akt and NF-kappaB signaling pathway.
Eur J Pharmacol. 642:146–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen J, Crawford R and Xiao Y: Vertical
inhibition of the PI3K/Akt/mTOR pathway for the treatment of
osteoarthritis. J Cell Biochem. 114:245–249. 2013. View Article : Google Scholar : PubMed/NCBI
|