Introduction
Diabetic cardiomyopathy (DCM), a severe and common
cardiac complication of diabetes, has become the leading cause of
mortality and morbidity in diabetic patients (1). Increasing evidence has suggested that
hyperglycemia, increased apoptosis, insulin
resistance/hyperinsulinemia and abnormal fatty acid metabolism are
the most important pathophysiological mechanisms of DCM (2–4). In
particular, hyperglycemia has generally been considered as a
central trigger in the pathophysiology of DCM, which leads to
increased oxidative stress through aggravating glucose oxidation
and production of reactive oxygen species (ROS), resulting in DNA
damage and accelerated apoptosis (5). Emerging evidence has also demonstrated
that diabetes may be caused by an imbalance of metabolic molecules,
including glucose, amino acids, lipids, and other such factors are
able to promote myocardial cells damage (6). Despite this, DCM remains poorly
understood and the underlying mechanisms are not completely
elucidated. A number of studies have established the cellular model
of DCM by using high D-glucose (HG) to simulate hyperglycemia in
DCM (7,8). Therefore, it is important to
investigate the molecular mechanism of HG-induced myocardial cell
injury, which may provide novel insights and therapeutic strategies
for the pathological process of DCM.
MicroRNAs (miRNAs or miRs) are a large class of
endogenous, small, noncoding RNAs that regulate diverse biological
processes including cell proliferation, cell differentiation,
apoptosis, and organ development (9). Emerging evidence reveals that changes
in the levels of miRNAs are associated with the occurrence and
development of various diseases, including diabetes and
cardiovascular diseases (10–12). In
recent years, the roles of miRNAs in DCM have received more
attention in research. A series of animal and cellular experiments
have identified changes in several miRNAs levels and their specific
mRNA targets which are associated with the pathogenetic processes
of diabetic heart complication (13,14).
Notably, in a previous study, the present authors determined that
several miRNAs are altered in DCM, as miR-106b-5p, −144-3p,
−186-5p, −22-3p and −30d-5p were downregulated, whereas
miR-516a-5p, −575, and −630 were upregulated in the serum of
patients with DCM (13). Of these
changes, the reduction of miR-186-5p was the most marked. In
addition, Bostjancic et al (15) have clarified that the dysregulation
of many miRNAs, such as miR-186, is believed to be associated with
a variety of physiological and pathological processes. Together,
these findings provide rationale for investigating the role of
miR-186-5p in the development of DCM, which will help identify the
molecular mechanisms and novel therapeutic strategies for DCM.
In the present study, AC16 cardiomyocytes were used
to assess the potential effect of miRNA-186-5p on HG-exerted damage
in the presence or absence of miRNA-186-5p mimic and miRNA-186-5p
inhibitor. To the best of our knowledge, the present results
confirmed for the first time that the downregulation of
miRNA-186-5p mediates HG-elicited cytotoxicity and apoptosis in
AC16 cardiomyocytes.
Materials and methods
Materials
Hoechst 33258, bicinchoninic acid (BCA) protein
assay kit, enhanced chemiluminescence (ECL) solution, and
radioimmunoprecipitation assay (RIPA) buffer were supplied by
Beyotime Institute of Biotechnology (Haimen, China). The cell
counting kit-8 (CCK-8) was obtained from Dojindo Molecular
Technologies, Inc. (Kumamoto, Japan). F12/Dulbecco's modified
Eagle's medium (DMEM-F12) and fetal bovine serum (FBS) were
purchased from HyClone (GE Healthcare Life Sciences, Logan, UT,
USA). Primary antibodies for caspase-3 (cat. no. 9662) and GAPDH
(cat. no. 5174) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA), and horseradish peroxidase (HRP)-conjugated
secondary antibodies (cat. no. 00001-9) were obtained from
ProteinTech Group, Inc. (Danvers, MA, USA). Annexin V/propidium
iodide (PI) apoptosis kit was supplied by BD Pharmingen (BD
Biosciences, San Jose, CA, USA). Lipofectamine RNAiMAX and Opti-MEM
medium were supplied by Invitrogen (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The miRcute miRNA Isolation kit, miRcute miRNA
First-strand cDNA Synthesis kit and miRcute miRNA quantitative
polymerase chain reaction (qPCR) detection kit were obtained from
Tiangen Biotech Co., Ltd. (Beijing, China).
Cell culture and treatments
The AC16 human adult ventricular cardiomyocyte cell
line was purchased from American Type Culture Collection (Manassas,
VA, USA) and cultured in DMEM-F12 medium with 10% FBS and 1%
penicillin-streptomycin at 37°C in a humidified incubator
containing 5% CO2. For HG treatment, AC16 cells grown to
~70% confluence were treated with HG (33 mM) for different
durations (0, 24, 48 or 72 h). For miR-186-5p mimic treatment,
cells were grown to 80–90% confluence and incubated at 37°C in
fresh, serum-free and antibiotic-free medium for 3 h, and then
transfected with miR-186-5p mimic (2 µg) or negative-control miRNA
(2 µg) for 5 h prior to incubation at 37°C in HG for 24 h. For
miR-186-5p inhibition treatment, cells were grown to 80–90%
confluence and transfected with miR-186-5p inhibitor (2 µg) for 5 h
and then incubated at 37°C with normal glucose (5.5 mM) for 24 h.
The sequence information on miR-186-5p mimics and inhibitors are as
follows: miR-186-5p mimics, 5′-CAAAGAAUUCUCCUUUUGGGCU-3′ and
miR-186-5p inhibitor, 5′-AGCCCAAAAGGAGAAUUCUUUG-3′.
MTS assay
AC16 cells were seeded on 96-well plates at
5×104 cells/well overnight at 37°C. Following treatment
of AC16 cells with miR-186-5p mimic or inhibitor in the presence
and absence of HG, respectively, for 24 h, and 10 µl MTS reagent
was added to each well for 1 h at 37°C. The absorbance values were
measured at 490 nm using a microplate spectrophotometer (Thermo
Fisher Scientific, Inc.). Results were expressed as a percentage of
control cells (transfected with negative-control miRNA). Each assay
was independently performed in triplicate. Appreciation rate (%) =
(mean OD value at time point/mean OD value at 0 Day-1) ×100.
Suppression rate (%)=(1-mean OD value of experimental group/control
group) ×100.
Hoechst 33258 staining
Nuclear morphology of AC16 cells was assessed via
Hoechst 33258 staining. Briefly, AC16 cells were seeded into a
24-well plate at a density of 1×105 cells/well
overnight. Following treated as described above for 24 h, AC16
cells were washed with PBS three times and fixed with 4%
formaldehyde for 10 min at 4°C. Cells were washed three times with
PBS again and incubated with 10 µg/ml Hoechst 33258 at room
temperature for 10 min in the dark. The morphological changes of
AC16 cells were observed at ×200 magnification under a fluorescence
microscope (Eclipse Ti; Nikon Corporation, Tokyo, Japan). Apoptosis
rate was calculated as the number of apoptotic cells/total cells
from the average of 5 random fields.
Annexin V-fluorescein isothiocyanate
(FITC) and PI staining
The apoptosis of AC16 cells was evaluated using an
Annexin V-PI double staining assay kit according to the
manufacturer's protocol. Briefly, AC16 cells were treated for 24 h
as detailed above and the culture supernatant was collected.
Subsequently, AC16 cells were washed and harvested with cold PBS.
AC16 cells and supernatant were co-centrifuged at 1,000 × g at room
temperature for 5 min. Following washing with PBS twice, cells were
resuspended in 500 µl binding buffer from the Annexin V-PI kit and
then co-incubated with 5 µl Annexin V-FITC reagent and 10 µl PI
reagent for 10 min at 4°C in the dark. Cellular apoptosis was
detected by flow cytometry (FC500; Beckman Coulter, Inc., Brea, CA,
USA).
Reverse transcription (RT)-qPCR
analysis
The level of miR-186-5p in AC16 cells, following
treatment as described above, was detected with RT-qPCR. Total RNA
was extracted from AC16 cells using an miRcute miRNA Isolation kit
according to manufacturer's protocol. Single-strand cDNA was
synthesized using an miRcute miRNA First-strand cDNA Synthesis kit
according to manufacturer's protocol. qPCR was performed via a one
step method with an miRcute miRNA qPCR Detection kit according to
the manufacturer's protocol. U6 small nuclear RNA was used as an
internal reference. Bulge-loop miRNA RT-qPCR Primer Sets (one RT
primer and a pair of qPCR primers for each set) specific for
miR-186-5p were designed by RiboBio (Guangzhou, China). Primer
sequence of miR-186-5p was as follows: forward,
5′-TCAAAGAATTCTCCTTTTGGGCT-3′ and reverse
5′-CGCTTCACGAATTTGCGTGTCAT-3′. PCR was performed for 2 min at 94°C,
followed by 40–45 cycles of 94°C for 20 sec and 60°C for 34 sec.
The amplified products were measured using 1% agarose gel
electrophoresis and densitometry of bands was analyzed using Image
J software bundled with Java 1.8.0–112 (National Institutes of
Health, Bethesda, MD, USA). Relative quantitative values were
calculated using the 2−ΔΔCq method (16).
Western blot analysis
AC16 cells were treated as detailed above and lysed
in RIPA buffer containing 1 mM phenylmethane sulfonyl fluoride
(Beyotime Institute of Biotechnology). Cellular protein was
centrifuged at 12,000 × g for 10 min at 4°C and quantified using a
BCA protein assay kit according to the manufacturer's protocol.
Equal amounts of proteins (50 µl) were separated on 12% SDS-PAGE
and electrotransferred onto a polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The membrane was then blocked with
5% no-fat milk in 0.05% Tween 20/TBS (TBST) for 2 h at room
temperature. Following washing three times with TBST, the membrane
was incubated with primary antibodies against cleaved caspase-3
(1:2,000) and GAPDH (1:2,000) overnight at 4°C. The membrane was
subsequently incubated with a HRP-conjugated secondary antibody
(1:5,000) for 2 h at room temperature and developed using an ECL
solution. GAPDH was used as the internal loading control. The band
densities were determined with Quantity One 4.6 software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Each assay was
independently performed in triplicate.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. The difference between groups was determined by one-way
analysis of variance followed by Fisher's least significance
difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
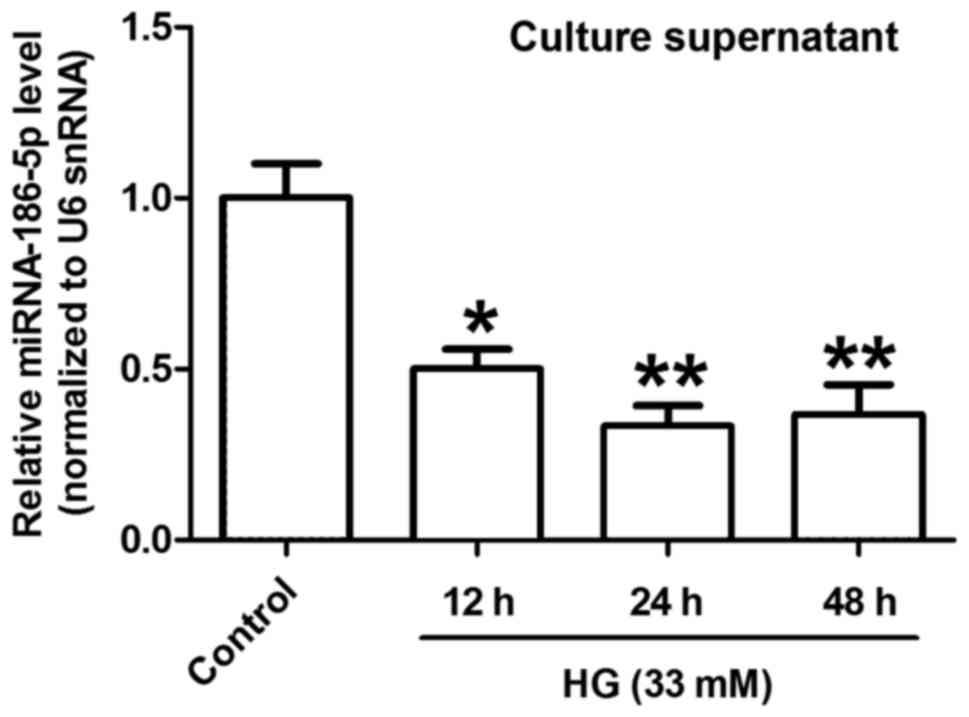
The level of miR-186-5p is
downregulated in HG-treated AC16 cardiomyocytes
RT-qPCR was performed to measure the change of
miR-186-5p level in HG-treated AC16 cardiomyocytes. As presented in
Fig. 1, HG treatment for 12, 24 and
48 h significantly decreased the level of miR-186-5p in AC16 cells
compared with control cells. The greatest decrease in miR-186-5p
was observed following 24 h treatment. Therefore, 24 h was
identified as the optimal treatment time for subsequent
experiments.
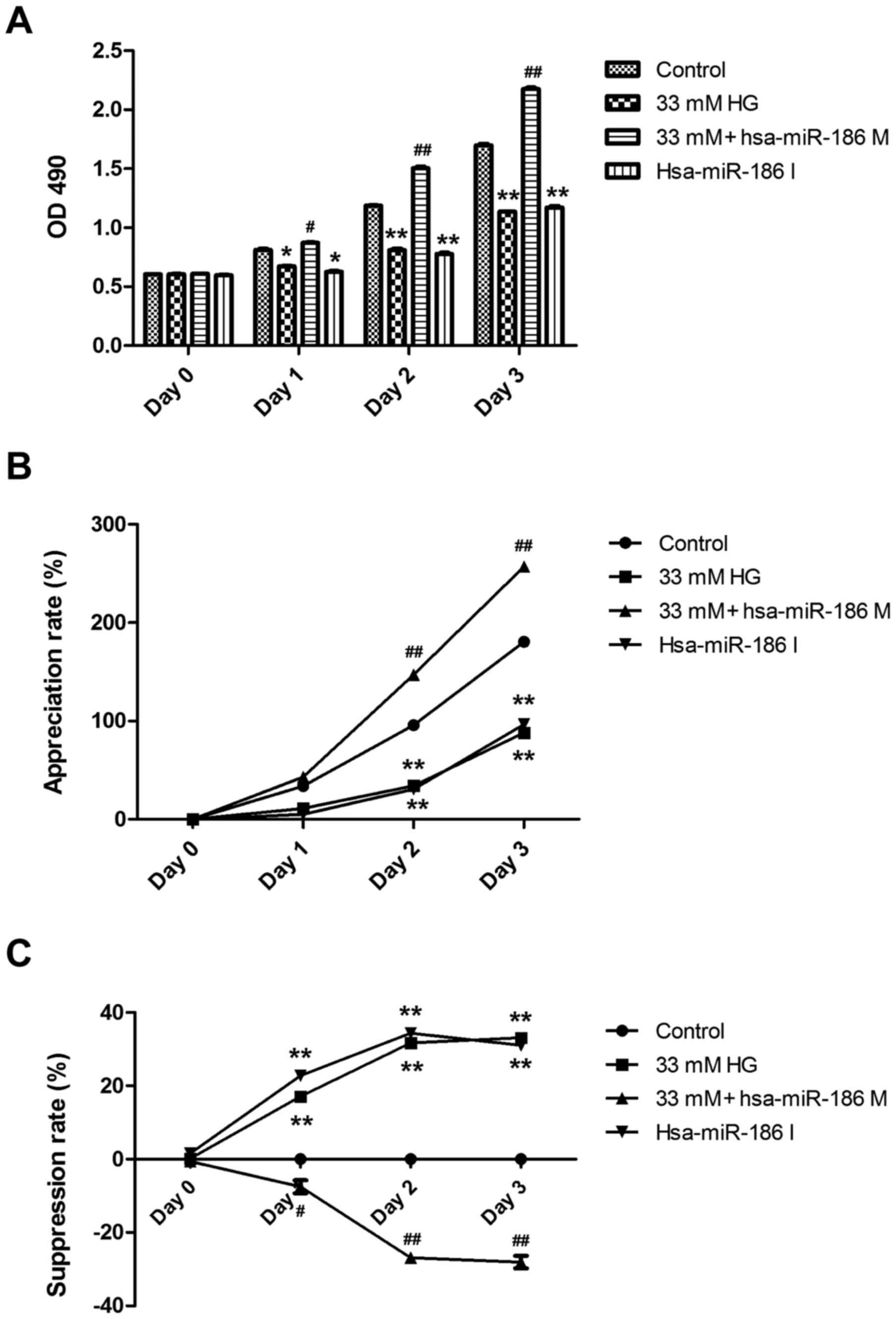
miR-186-5p mimic transfection reverses
HG-induced downregulation of cell viability in AC16
cardiomyocytes
To investigate whether miR-186-5p is associated with
HG-induced myocardial cytotoxicity, we detected the effect of
miR-186-6p mimic, which is able to simulate the high level of
mature miR-186-6p in cells, or miR-186-5p inhibitor on the
viability of AC16 cells. As presented in Fig. 2, pre-transfection of AC16 cells with
miR-186-5p mimic significantly reversed the downregulation of cell
viability induced by HG (Fig. 2A).
In addition, HG induced a significant decrease in the appreciation
rate of AC16 cells, compared with controls (Fig. 2B) and a significant increase in the
suppression rate of AC16 cells (Fig.
2C), which were both significantly ameliorated by miR-186-5p
mimic, which suggests that the AC16 cells transfected with
miR-186-5p mimic had the ability to protect against long-term
HG-induced injury. Furthermore, it was demonstrated that miR-186-5p
inhibitor significantly downregulated the viability and
appreciation rate (Fig. 2A and B)
and significantly upregulated the suppression rate of AC16 cells
(Fig. 2C) compared with controls,
similar to treatment with HG, which suggests that miR-186-5p may
have an important role in maintaining cell survival. These results
showed that HG was able to induce cytotoxicity through
downregulating the level of miR-186-5p in AC16 cells.
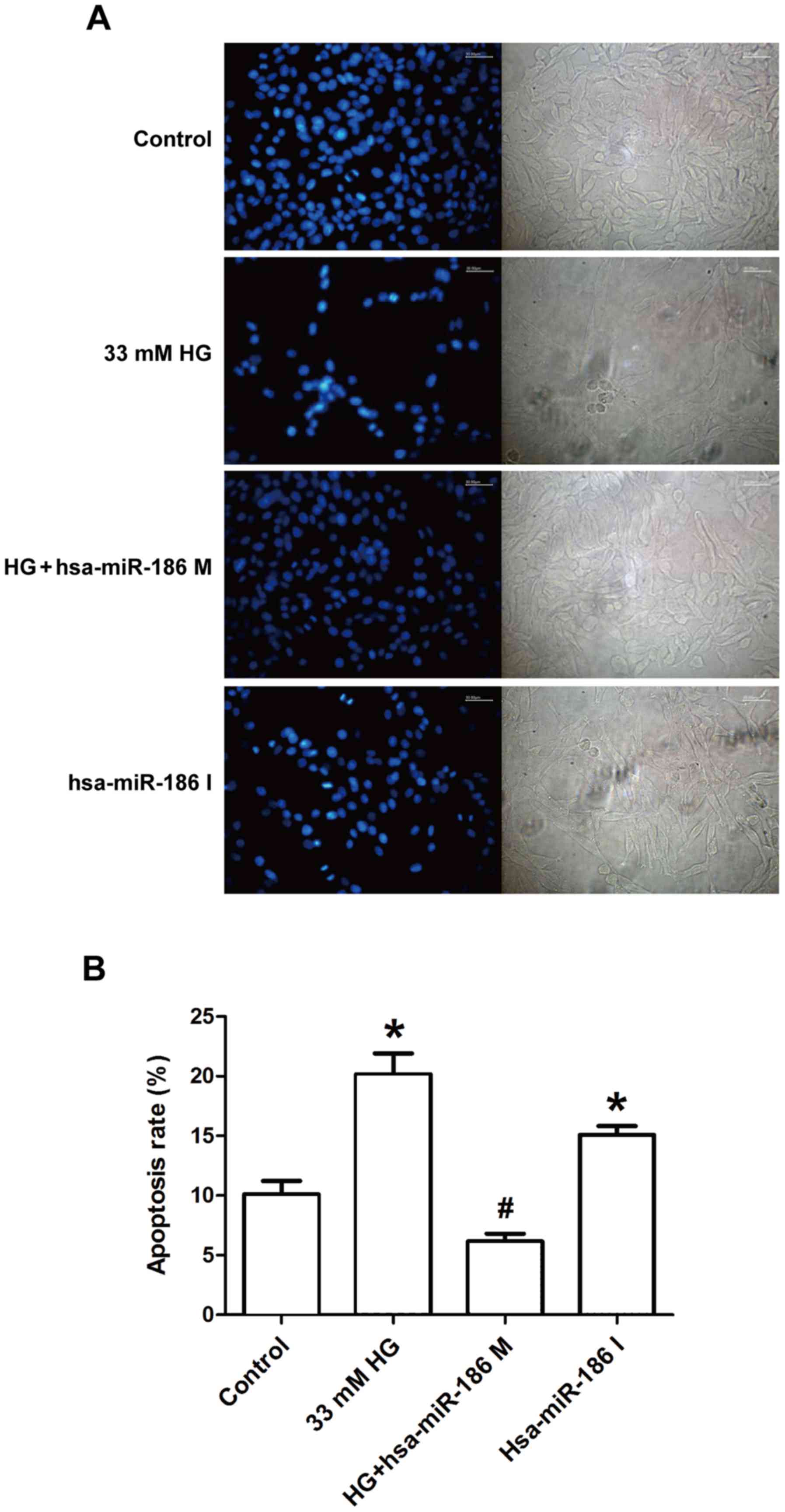
miR-186-5p mimic ameliorates
HG-induced morphological changes of AC16 apoptotic cells
To determine whether an association exists between
HG-induced cardiomyocyte apoptosis and miR-186-5p, the effect of
miR-186-5p mimic and miR-186-5p inhibitor was observed on apoptosis
in AC16 cells via Hoechst 33258 staining. As shown in Fig. 3, in the control group, AC16 cells
exhibited regular-shaped nuclei and low intensity blue uniform
fluorescence, whereas the numbers of AC16 cells with fragmented or
condensed nuclei and bright blue fluorescence, which were
characteristics of apoptotic cells. were markedly increased in
HG-treated cells (Fig. 3A). However,
compared with HG group, the number of AC16 cells with bright blue
fluorescence was markedly decreased in the miR-186-5p mimic group.
Furthermore, the rate of apoptosis was significantly upregulated by
HG, in comparison with control cells; however, this was
significantly ameliorated in miR-186-5p mimic cells (Fig. 3B). In addition, transfection with
miR-186-5p inhibitor led to fragmented or condensed nuclei in AC16
cells. These results suggest that HG downregulated the level of
miR-186-5p in AC16 cells, and therefore induced apoptosis.
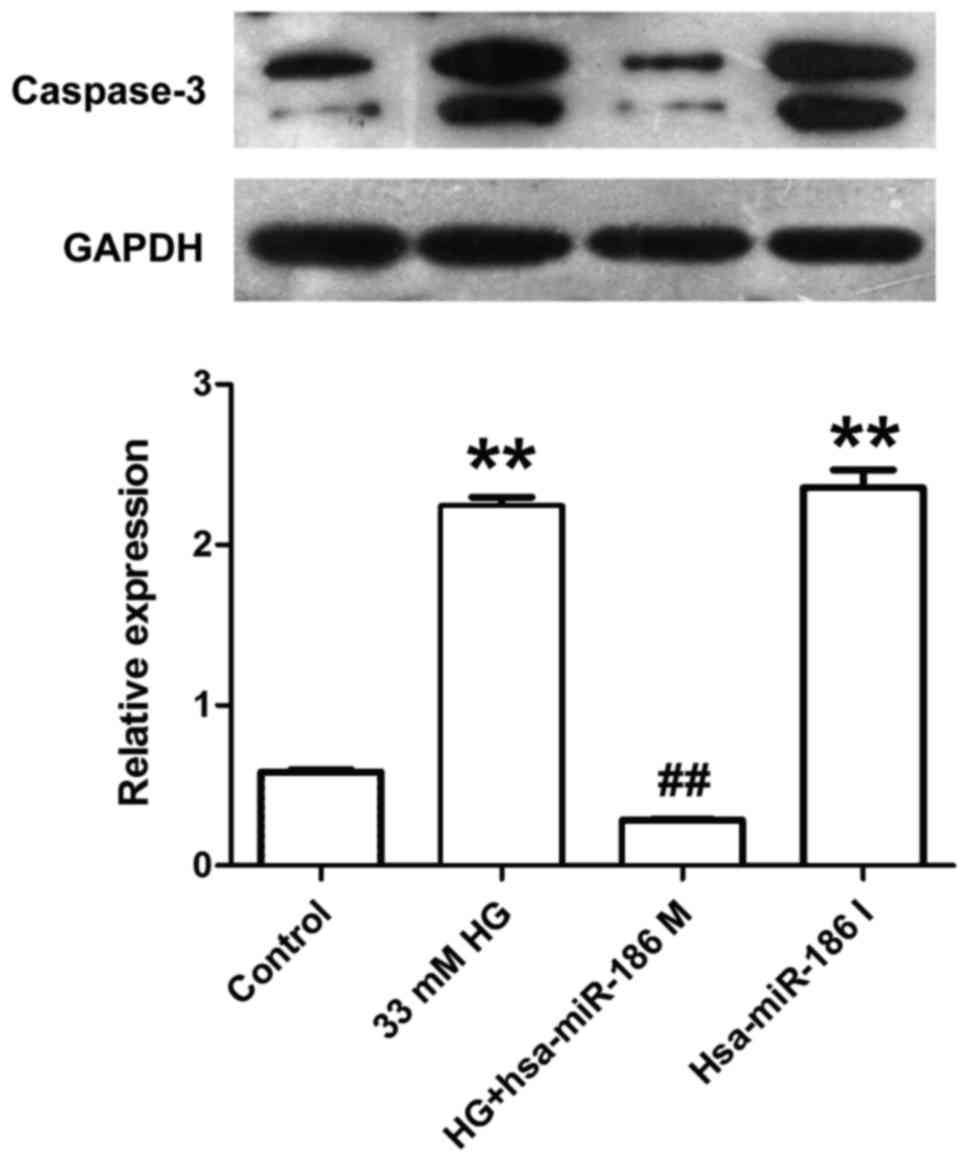
miR-186-5p mimic alleviates HG-induced
increase in the expression of cleaved caspase-3 protein in AC16
cardiomyocytes
Caspase-3 is considered as a key effector protease
in the cellular apoptotic response, which is activated in the
process of apoptosis followed by substrate binding, resulting in
cell apoptosis through amplifying the cascade reaction (17). Therefore, the effect of miR-186-5p on
cleaved caspase-3 level was measured in AC16 cells. As presented in
Fig. 4, AC16 cells treated with HG
or transfected with miR-186-5p inhibitor exhibited a significantly
increased expression of cleaved caspase-3 protein compared with
that of the control group. However, compared with the HG group,
transfection with miR-186-5p mimic significantly decreased the
expression of cleaved caspase-3 protein in AC16 cells. This
suggests that HG induced an upregulation of cleaved caspase-3
protein expression via reducing the miR-186-5p level.
miR-186-5p mimic ameliorates
HG-induced apoptosis in AC16 cardiomyocytes
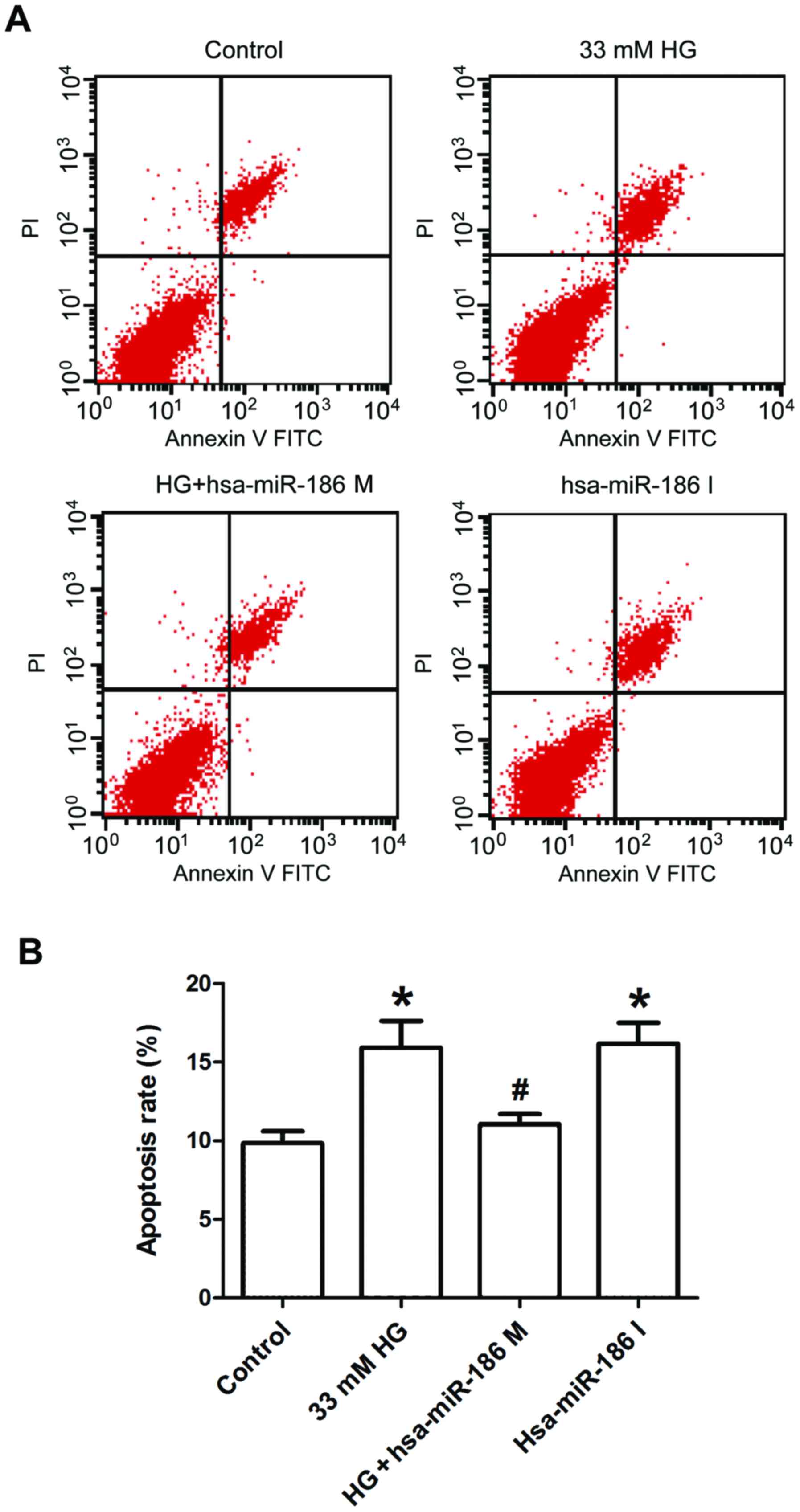
Finally, Annexin V/PI staining and flow cytometry
were used to further test the effect of miR-186-5p on apoptosis in
AC16 cells. As presented in Fig. 5,
AC16 cells treated with HG or transfected with miR-186-6p inhibitor
significantly increased the percentage of apoptotic cells in
comparison with the control group, whereas transfection with
miR-186-5p mimic significantly ameliorated the HG-induced increase
in the apoptotic ratio. This result suggested that HG induced
apoptosis through downregulating the miR-186-5p level in AC16
cells.
Discussion
Diabetes is associated with hyperglycemia, insulin
resistance and dyslipidemia, which are risk factors for
cardiovascular diseases. The prevalence of cardiac diabetic
diseases such as DCM has been increasing worldwide, and is the
leading cause of morbidity and mortality among diabetic patients
(18). However, the underlying
mechanisms of DCM remain to be elucidated. In recent years, the
role of miRNAs in the development of DCM has been an area of
interest for research (13,19). Therefore, the aim of the present
study was to investigate whether changes in miR-186-5p expression
are associated with HG-induced AC16 cardiomyocyte injury, which is
an in vitro cellular model of hyperglycemia-induced
myocardial injury (20).
miRNAs have been demonstrated to have important
roles in various forms of cardiovascular disease. Bostjancic et
al (15) used miRNA microarrays
to screen the differential expression of miRNA in human myocardial
infarction and found that miRNA-186 was dysregulated under
myocardial infarction. Consistent with this finding, the present
study also demonstrated that miR-186-5p was downregulated in
HG-treated AC16 cells, indicating the potential mechanism for
expanding the therapeutic strategies. There is also evidence that
miR-186 participates in modulating glucose uptake as well as
activating cell cycle checkpoint under disease conditions (21). In the present study, HG-induced
injury resulted in the decreased viability and appreciation rate as
well as increased suppression ratio of AC16 cardiomyocytes, which
is consistent with previous findings. These findings suggest that
the (22) decreased level of
miR-186-5p contributes to HG-induced cell damage.
Accumulating evidence demonstrates that myocardial
cell death is considered as a major event in the progression of
cardiovascular diseases, and suppression of myocardial cell death
for apoptosis-specific signaling pathways results in a significant
prevention of DCM (23,24). The activity of caspase-3 and
apoptosis were markedly increased in a mouse model of
streptozotocin-induced DCM (25) and
in hyperglycemia-induced H9c2 cardiac myoblasts (26). In addition, there is also evidence
that miR-186 transfection induced apoptosis, whereas anti-miR-186
transfection reduced apoptosis under disease conditions (27,28),
indicating that miR-186 promotes apoptosis. Notably, Zhang et
al (29) investigated the
effects of miRNA-186 overexpression or inhibition on apoptosis in
A549 cells, and demonstrated that the significant downregulation of
miRNA-186 expression was associated with curcumin-induced
apoptosis. Based on these research findings, it can be determined
that miR-186 has a complex association with apoptosis and the
varying roles of miR-186 in apoptosis may be associated with the
regulation of different downstream signaling pathways or different
subtypes. In the present study, whether changes in miR-186 level
were associated with HG-induced apoptosis was also investigated. it
was demonstrated that miR-186-5p mimic downregulated the expression
of caspase-3 protein, whereas miR-186-5p inhibitor significantly
upregulated the expression of caspase-3 protein in AC16
cardiomyocytes, which was consistent with the observation
delineated by Sha et al (30). In addition, morphological changes of
apoptotic cells induced by HG were ameliorated by miR-186-5p mimic
transfection. The HG-induced upregulation of apoptosis rate was
also ameliorated by miR-186-5p mimic, which suggests that the
downregulation of miR-186-5p is associated with HG-induced
apoptosis, likely through activation of caspase-3. However, a
limitation of the present study is that the downstream target(s) of
miRNA-186 were not further explored. In addition, performing in
vivo experiments is necessary to elucidate the underlying
molecular mechanism of this phenomenon.
In conclusion, the present study demonstrated that
miR-186-5p was downregulated in HG-treated AC16 cells, miR-186-5p
mimic reversed HG-exhibited cytotoxicity and apoptosis and
miR-186-5p inhibitor increased apoptosis, which was the same effect
as HG in AC16 cells. These findings suggest that miR-186-5p
deletion ameliorates HG-induced injury, likely by modestly
promoting apoptosis, which may be a potential mechanism for
expanding the therapeutic strategies of DCM.
Acknowledgements
The present study was supported by Guangdong Natural
Science Foundation (grant nos. 2015A030310359 and S2011010002620)
and Science and Technology Planning Project of Guangdong in China
(grant no. 2012A080202020).
References
|
1
|
Acar E, Ural D, Bildirici U, Sahin T and
Yilmaz I: Diabetic cardiomyopathy. Anadolu Kardiyol Derg.
11:732–737. 2011.PubMed/NCBI
|
|
2
|
Bertoni AG, Tsai A, Kasper EK and Brancati
FL: Diabetes and idiopathic cardiomyopathy: A nationwide
case-control study. Diabetes Care. 26:2791–2795. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kain V and Halade GV: Metabolic and
biochemical stressors in diabetic cardiomyopathy. Front Cardiovasc
Med. 4:312017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bugger H and Abel ED: Molecular mechanisms
of diabetic cardiomyopathy. Diabetologia. 57:660–671. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Isfort M, Stevens SC, Schaffer S, Jong CJ
and Wold LE: Metabolic dysfunction in diabetic cardiomyopathy.
Heart Fail Rev. 19:35–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ramasarma T and Rafi M: A glucose-centric
perspective of hyperglycemia. Indian J Exp Biol. 54:83–99.
2016.PubMed/NCBI
|
|
8
|
Feuvray D: Diabetic cardiomyopathy. Arch
Mal Coeur Vaiss. 97:261–265. 2004.PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vickers KC, Rye KA and Tabet F: MicroRNAs
in the onset and development of cardiovascular disease. Clin Sci
(Lond). 126:183–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McClelland AD and Kantharidis P: microRNA
in the development of diabetic complications. Clin Sci (Lond).
126:95–110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tyagi AC, Sen U and Mishra PK: Synergy of
microRNA and stem cell: A novel therapeutic approach for diabetes
mellitus and cardiovascular diseases. Curr Diabetes Rev. 7:367–376.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
León LE, Rani S, Fernandez M, Larico M and
Calligaris SD: Subclinical detection of diabetic cardiomyopathy
with MicroRNAs: Challenges and perspectives. J Diabetes Res.
2016:61431292016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Asrih M and Steffens S: Emerging role of
epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc
Pathol. 22:117–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bostjancic E, Zidar N and Glavac D:
MicroRNA microarray expression profiling in human myocardial
infarction. Dis Markers. 27:255–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Juraver-Geslin HA and Durand BC: Early
development of the neural plate: New roles for apoptosis and for
one of its main effectors caspase-3. Genesis. 53:203–224. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yilmaz S, Canpolat U, Aydogdu S and Abboud
HE: Diabetic cardiomyopathy; summary of 41 years. Korean Circ J.
45:266–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X and Liu S: Role of microRNAs in the
pathogenesis of diabetic cardiomyopathy. Biomed Rep. 6:140–145.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
You Q, Wu Z, Wu B, Liu C, Huang R, Yang L,
Guo R, Wu K and Chen J: Naringin protects cardiomyocytes against
hyperglycemia-induced injuries in vitro and in vivo. J Endocrinol.
230:197–214. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun P, Hu JW, Xiong WJ and Mi J: miR-186
regulates glycolysis through Glut1 during the formation of
cancer-associated fibroblasts. Asian Pac J Cancer Prev.
15:4245–4250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang JL, Xiao DZ, Liu XY, Lin QX, Shan
ZX, Zhu JN, Lin SG and Yu XY: High glucose induces apoptosis in
AC16 human cardiomyocytes via macrophage migration inhibitory
factor and c-Jun N-terminal kinase. Clin Exp Pharmacol Physiol.
37:969–973. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee Y and Gustafsson AB: Role of apoptosis
in cardiovascular disease. Apoptosis. 14:536–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai L and Kang YJ: Cell death and diabetic
cardiomyopathy. Cardiovasc Toxicol. 3:219–228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G,
Chen R, Li J, Fan GC, Lacefield JC and Peng T: Silencing of miR-195
reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia.
58:1949–1958. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun X, Chen RC, Yang ZH, Sun GB, Wang M,
Ma XJ, Yang LJ and Sun XB: Taxifolin prevents diabetic
cardiomyopathy in vivo and in vitro by inhibition of oxidative
stress and cell apoptosis. Food Chem Toxicol. 63:221–232. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun KX, Jiao JW, Chen S, Liu BL and Zhao
Y: MicroRNA-186 induces sensitivity of ovarian cancer cells to
paclitaxel and cisplatin by targeting ABCB1. J Ovarian Res.
8:802015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He W, Feng J, Zhang Y, Wang Y, Zang W and
Zhao G: microRNA-186 inhibits cell proliferation and induces
apoptosis in human esophageal squamous cell carcinoma by targeting
SKP2. Lab Invest. 96:317–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Du Y, Wu C, Ren X, Ti X, Shi J,
Zhao F and Yin H: Curcumin promotes apoptosis in human lung
adenocarcinoma cells through miR-186* signaling pathway. Oncol Rep.
24:1217–1223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sha WG, Shen L, Zhou L, Xu DY and Lu GY:
Down-regulation of miR-186 contributes to podocytes apoptosis in
membranous nephropathy. Biomed Pharmacother. 75:179–184. 2015.
View Article : Google Scholar : PubMed/NCBI
|