Introduction
Primary hyperoxaluria is a rare autosomal recessive
disorder of glyoxylate metabolism in which specific deficiencies of
hepatic enzymes cause excessive oxalate production (1). Primary hyperoxaluria accounts for ~10%
of pediatric patients with nephrocalcinosis (2) and 2% of patients undergoing renal
replacement therapy (3). There are
three known types of primary hyperoxaluria; however, the mechanism
involved in manifestation are unknown (4). These known types of primary
hyperoxaluria are caused by deficiencies of AGXT, GRHPR and HOGA1,
which encode alanine, glyoxylate aminotransferase,
reductase/hydroxypyruvate reductase and hydroxyoxoglutarate
aldolase, respectively (5). GPHPR,
which is defective in primary hyperoxaluria type 2, catalyzes the
conversion of hydroxypyruvate to d-glycerate (6). Primary hyperoxaluria type 2 is
characterized by recurrent episodes of nephrolithiasis and
nephrocalcinosis (7). In some cases,
primary hyperoxaluria type 2 may result in end-stage renal disease;
however, the prevalence of this is low (8). Conservative measures, including
adequate hydration and oral citrate supplementation, are essential
to preserve renal function and prevent nephrolithiasis for patients
with early hyperoxaluria (4).
Molecular targeted therapy is still currently being explored in
cell systems and animal models, but has not been fully investigated
in humans (9).
The present study described a case of primary
hyperoxaluria type 2 with multiple bouts of nephrolithiasis
resistant to lithotripsy and conservative therapy, which eventually
progressed to chronic renal failure requiring kidney
transplantation.
Case report
A 33-year old man was admitted to our hospital
(Transplant Center, First Hospital of Jilin University, Changchun,
China) in November 2014 because of chronic renal failure for 2
years. He was diagnosed with nephrolithiasis in March 2004, at
which time a single stone in the right kidney was identified and
removed by lithotripsy. Multiple renal calculi in bilateral kidneys
were identified in June, 2009 and managed via lithotripsy and the
patient received Quercus salicina extract capsules (450 mg;
three times daily for 3 weeks). However, bilateral renal calculi
recurred 2 months after the lithotripsy, and the patient underwent
multiple lithotripsies thereafter. In November 2014, the patient
developed dizziness and nausea and his serum creatine levels were
measured as 2,500 mol/l (normal reference values, 44 to 115
µmol/l). The patient was diagnosed with chronic renal failure and
underwent maintenance dialysis.
The patient received an allogeneic renal transplant
in December 2014 at our Transplant Center. The Transplant was
performed in accordance with the approved guidelines by the Ethics
Committee of Transplant Center, First Hospital of Jilin University
and the patient provided signed and informed consent. Urine was
passed 30 sec following the restoration of blood flow. The patient
was administered intravenous anti-human T thymocyte rabbit
immunoglobulin (100 mg once daily for 6 days; Fresenius SE &
Co. KGaA, Bad Homburg, Germany) and methylprednisolone (500 mg once
daily for 3 days; Pfizer, Inc., Reno, NV, USA) and oral tacrolimus
(0.1 mg/kg/day; Astellas Ireland Co., Ltd., Kerry, Ireland),
mycophenolate (540 mg twice daily; Novartis, Basel, Switzerland),
and prednisone (120 mg; Xianju Pharmaceuticals Co., Ltd., Hangzhou,
Zhejiang, China) on post-transplant day 3 when intravenous
methylprednisolone was completed. Oral prednisone was then reduced
20 mg/day until the dosage was 20 mg. The 24-h urine volume was
recorded as 11,350 ml on day 1 post-surgery. Serum creatine (normal
reference values, 44 to 115 mol/l) decreased to 198 µmol/l on day 5
post-surgery and rose to 214 mol/l on day 6 post-surgery. The
patient's blood concentration of tacrolimus was 9.1 ng/ml on day 8
post-surgery and a renal graft biopsy was performed. Biopsy tissues
were fixed in 10% formaldehyde at room temperature for 4 h and
sectioned at a thickness of 2 µm. Following standard protocols,
sections were processed and stained with hematoxylin and eosin
(H&E) staining. Briefly, the sections were stained in
hematoxylin at room temperature for 10 min, followed by staining in
1% eosin solution at room temperature for 3 min, and subsequently
washed with distilled water. The sections were then placed into
Schiff's reagent and incubated for 30 min at room temperature. The
slides were observed under light microscope (BX5; Olympus Corp.,
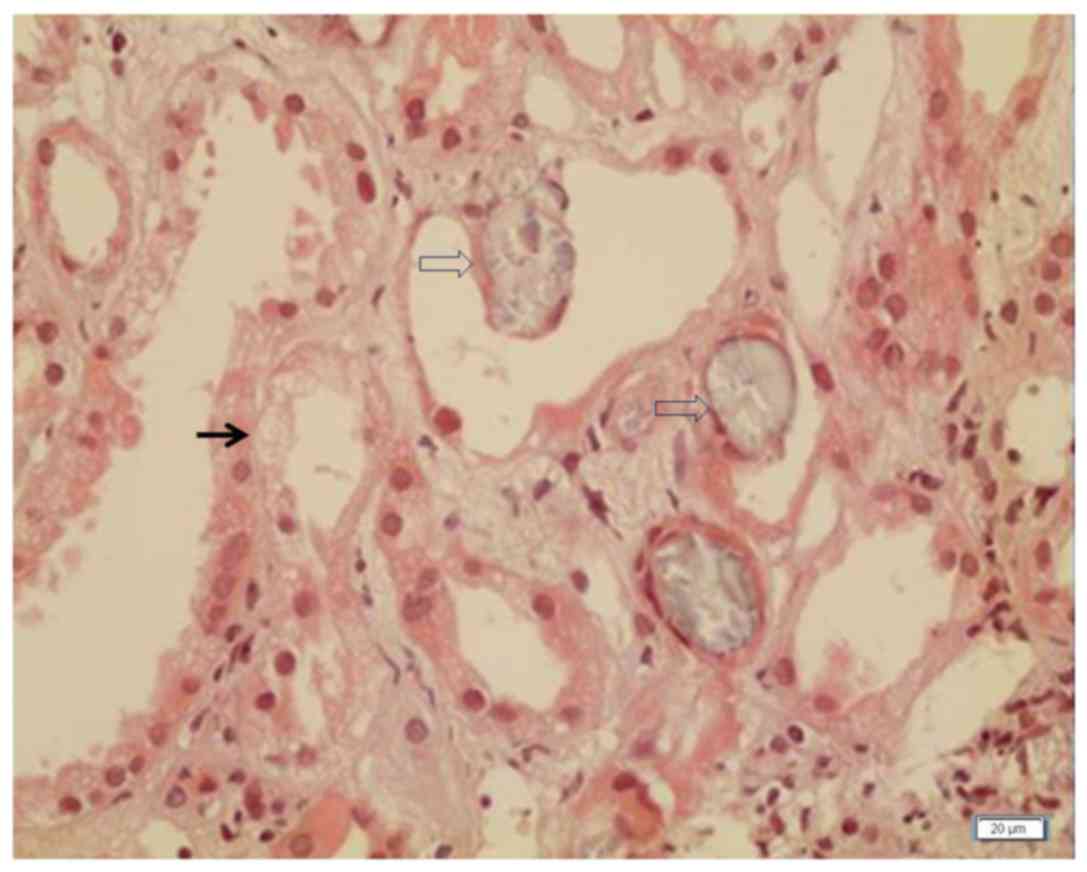
Tokyo, Japan) and mild acute nephrotoxicity was observed (Fig. 1). The dose of tacrolimus was reduced
to 0.075 mg/kg/day; however, no decline in serum creatine was
observed. On day 15 post-surgery, serum creatine levels gradually
increased to 291 µmol/l. A renal graft ultrasound found
hyperechocity of the renal cortex, and the transplanted kidney was
11.5×5.6×5.5 cm in size, with a renal artery resistive index of
0.65 (normal range, 0.6–0.8). The tacrolimus dose was further
reduced to 0.05 mg/kg/day and 1.0 mg/day sirolimus (Pfizer Ireland
Pharmaceuticals, Kildare, Ireland) was added. Blood rapamycin
content was 6.1 ng/ml, tacrolimus 5.1 ng/ml, and the area under the
curve (AUC) of mycophenolate mofetil was 33.38.
ELISA-plasma activity test for human leukocyte agent
class II was performed using an ELISA kit (LATM10X5, LOT001; One
Lambda; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according
to the manufacturer's instructions and the results were negative.
There was no noticeable improvement in renal function, and serum
creatine was 290 µmol/l. A renal biopsy was performed on day 26
post-surgery and the sections were processed and stained with
periodic acid-Schiff (PAS) stain following standard protocol. The
sections were immersed in 1% periodic acid at room temperature for
15 min, and subsequently washed with distilled water before the
sections were placed into Schiff's reagent and incubated 30 min at
room temperature. The slides were observed under light microscope
(BX51; Olympus Corp), which revealed borderline lesions of the
renal graft and tubulitis (1+) that was accompanied by a small
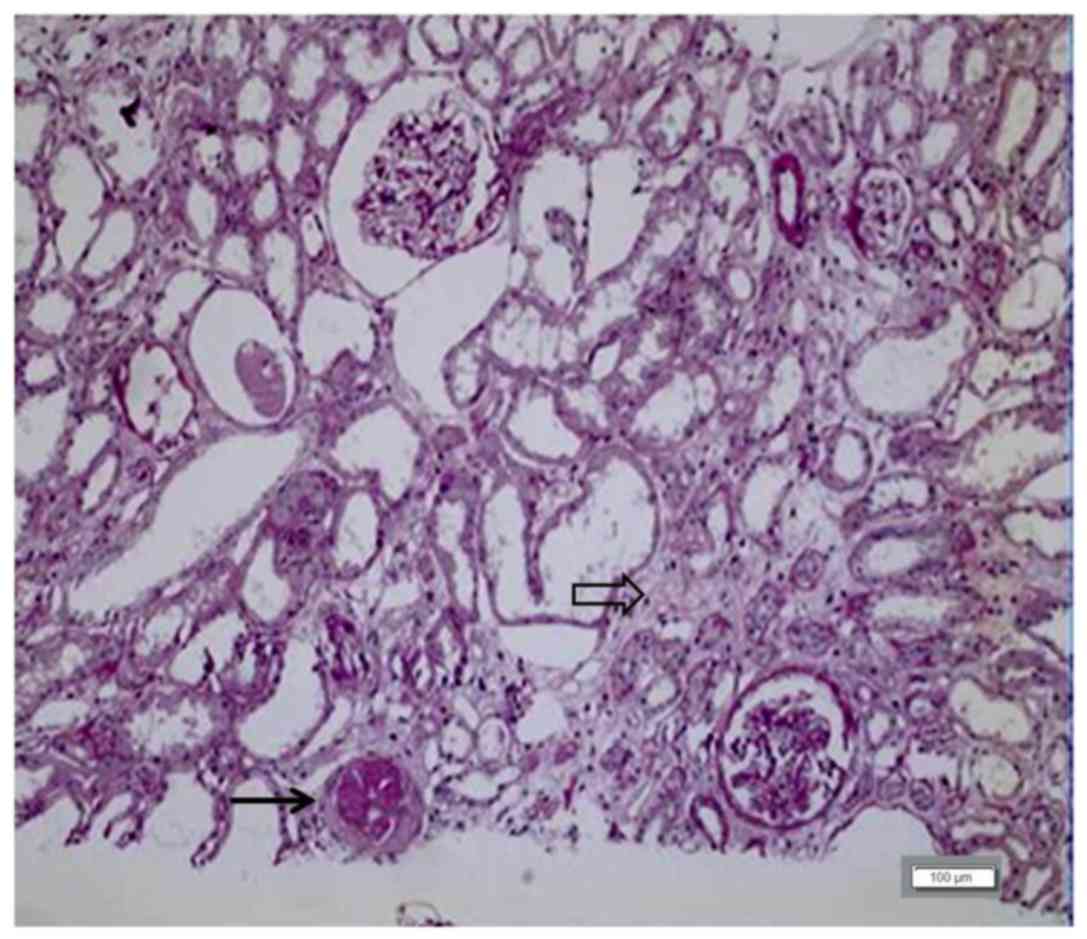
amount of crystal deposition within the renal tubules (Fig. 2). Primary hyperoxaluria was
considered. The patient was administered with sodium bicarbonate
tablets (1.0 g 3 times daily), oral vitamin B6 (100 mg twice
daily), and Quercus salicina extract capsules (450 mg 3
times daily) for discharging stones; however, no improvement was
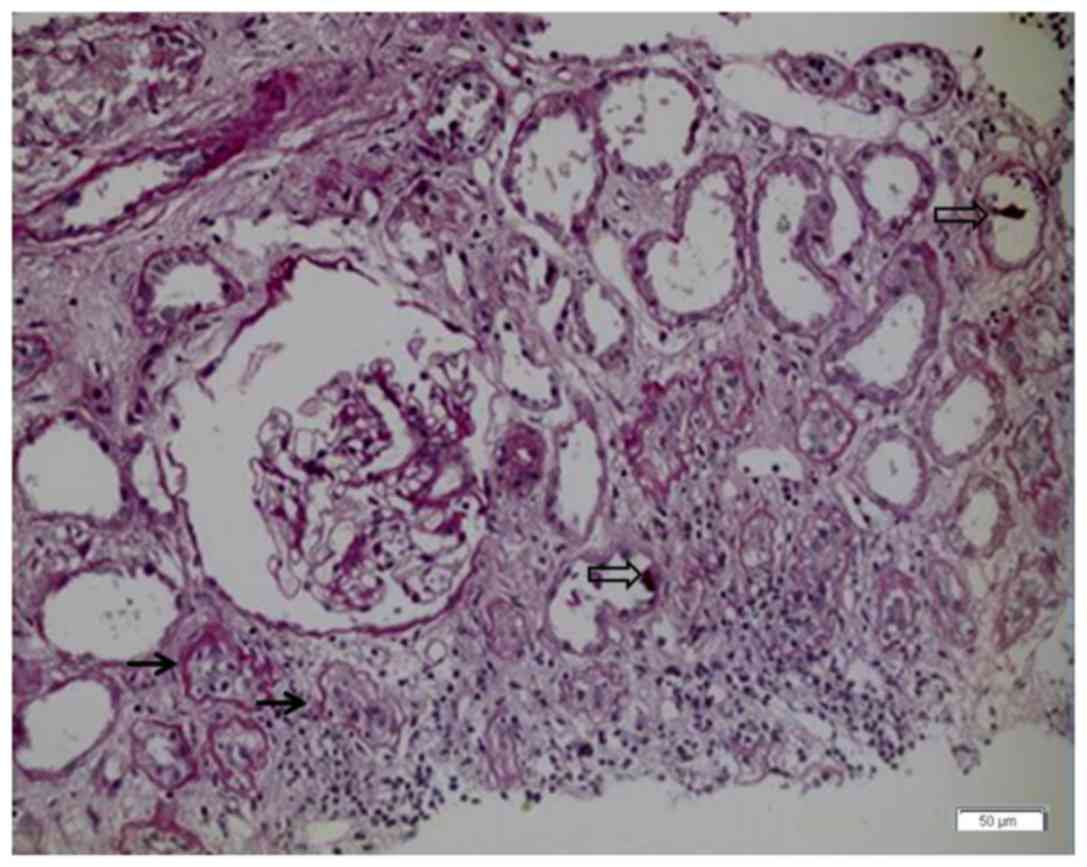
seen. Serum creatine rose to 376 µmol/l, and a protocol biopsy on
day 37 post-surgery revealed borderline lesions of the renal graft,
tubulitis (1+), and crystal deposition within the renal tubules,
which was worse than that on day 26 (Fig. 3).
A peripheral blood sample was taken from the patient
and genomic DNA was extracted using the TIANamp Blood DNA kit
(DP348-02 DP348-03; Tiangen Biotech, Co., Ltd., Beijing, China) as
instructed by the manufacturer. Primers covering all coding regions
and flanking introns of the Ph1 and Ph2, AGXT and
GRHPR genes were designed and synthesized (Qingwei, Wuhan,
China) according to the gene sequences from Ensembl (http://asia.ensembl.org/index.html) via NCBI
Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast)
(Primer sequences are available upon request). The gene sequences
were amplified by polymerase chain reaction (PCR) (2X PCR MasterMix
polymerase; Tiangen Biotech, Co., Ltd., Beijing, China). The PCR
conditions were as follows: 95°C for 5 min followed by 95°C for 30
sec; 56°C for 30 sec; 72°C for 30 sec for a total of 35 cycles and
an additional incubation at 72°C for 10 min. PCR products were
purified and sent to Grandomics Biosciences Co., Ltd. (Beijing,
China) for sequencing analysis. The sequencing results showed that
the patient was negative for Ph1 and Ph2.
Furthermore, no mutations were detected in the AGXT exon
coding region. However, a homozygous mutation was detected in the
GRHPR gene, and a heterozygous mutation in the gene was
detected in the patient's mother.
The patient's urine volume gradually declined and
dialysis was maintained at day 42 post-transplant. Due to repeated
episodes of fever, nausea, fatigue, and viral infections, the
patient requested that the renal transplant be removed. In February
2015 at day 54 post-transplant, the patient underwent a second
surgery under general anesthesia. An incision was made in the
lateral border of the rectus abdominis and the graft renal artery
and vein were located, ligated and excised. The ureter was also
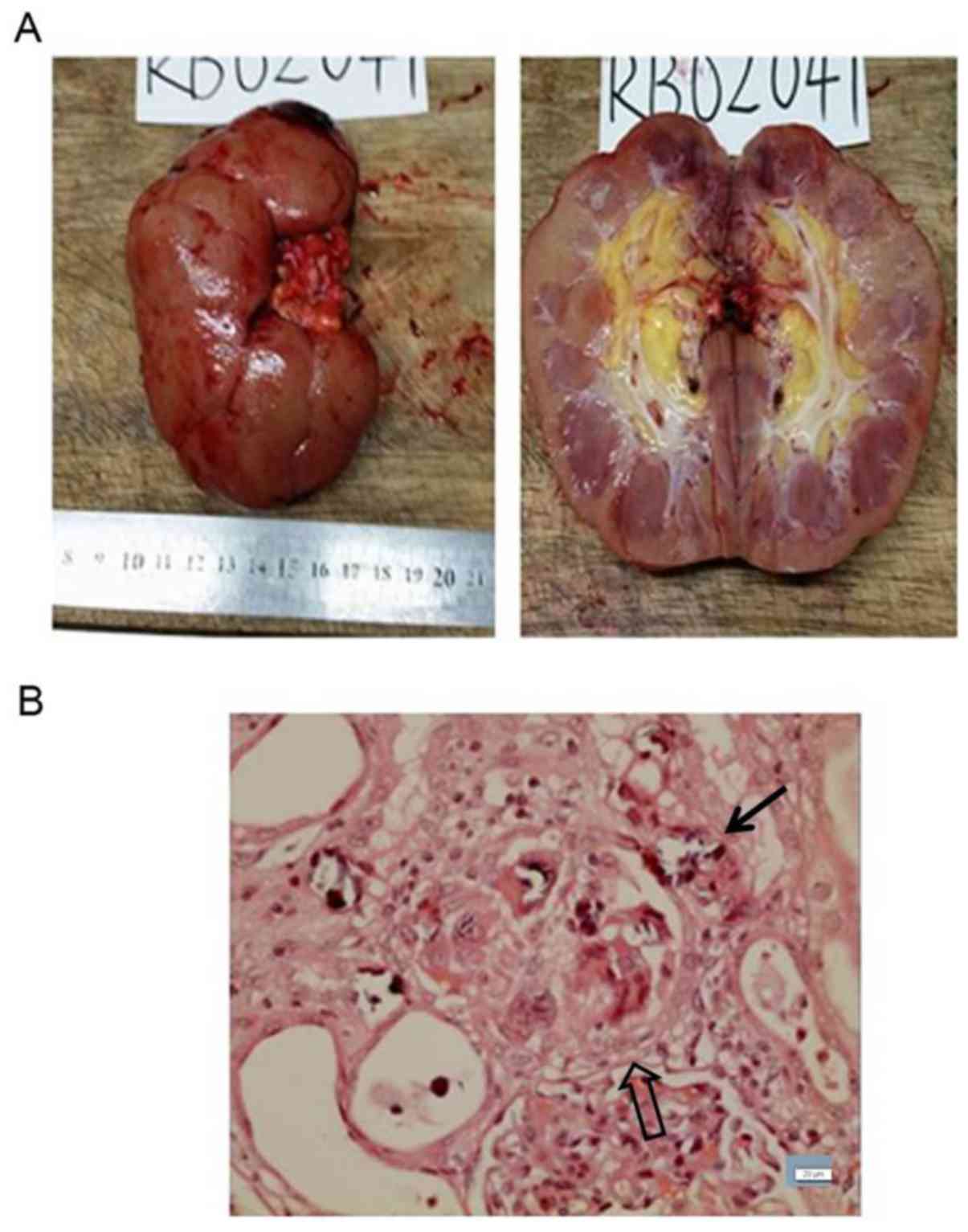
ligated and excised and the renal graft was removed. Renal
pathology revealed interstitial injury due to crystal deposition
within the renal tubules, with renal pelvis stones (Fig. 4). Chemical analysis found that the
kidney stones were calcium oxalate. The patient received
post-transplant hemodialysis and the date of the last follow-up
visit was October 2015.
Discussion
Primary hyperoxaluria, a rare autosomal recessive
disorder, is characterized by hyperoxaluria accompanied by early
and recurrent episodes of nephrolithiasis, which eventually causes
renal injury as a result of calcium oxalate crystal deposition
(7). Due to its rarity, physicians
are typically unfamiliar with the condition, resulting in delayed
or missed diagnosis (10). The
present patient suffered multiple bouts of nephrolithiasis
recalcitrant to lithotripsy and conservative therapy, which
eventually progressed to chronic renal failure, leading to
allograft transplantation. Undiagnosed primary hyperoxaluria also
contributed to the eventual failure of the renal graft. Two
protocol biopsies failed to reveal crystal deposition in the renal
tubules. Primary hyperoxaluria type 2 was only diagnosed following
the detection of crystal depositions in the renal tubules and
molecular genetic testing.
There are three types of primary hyperoxaluria. Type
1 is caused by a deficiency of the liver peroxisomal enzyme
alanine-glyoxylate-aminotransferase (AGT), which catalyzes the
conversion of glyoxylate to glycine (11). Type 2 is caused by a GPHPR
deficiency, which catalyzes the conversion of hydroxypyruvate to
d-glycerate (6). An enzyme
deficiency in type 3 has not been unambiguously identified and
mutations in the DHDPSL gene have been reported (12,13).
Primary hyperoxaluria is typically associated with early onset of
symptoms, presenting in patients between 1 and 25 years of age
(14). In the present case, the
patient was diagnosed in adulthood; however, he suffered symptoms
early in life. The findings from 24-h urine oxalate, urinary
oxalate-to-creatinine molar ratio and plasma oxalate suggested
primary hyperoxaluria type 1 (15),
as did hepatic AGT activity (16);
however, the final diagnosis was dependent on molecular genetic
testing (12,17). As primary hyperoxaluria 2 exhibits a
similar phenotype to type 1, its ultimate diagnosis relies on the
conclusion of molecular genetic testing (13). When primary hyperoxaluria is
suspected gene testing for type 1 should be performed first as it
is more common (18).
Primary hyperoxaluria is managed by ensuring that
the patient has adequate fluid intake, is given urinary inhibitors
of calcium oxalate crystallization and vitamin B6, and undergoes
routine dialysis (4). In this
patient, these conservative measures failed to control unrelenting
bouts of nephrolithiasis and nephrocalcinosis. Kidney
transplantation alone has been used to treat primary hyperoxaluria
with varied results (19,20). In cases of hyperoxaluria type 1,
although the renal graft is able to function, excessive oxalate
production occurs in the liver, which continues to deposit calcium
oxalate in the renal parenchyma and tubules (21). Therefore, renal transplantation is
not recommended for type 1 patients (21). In the present case, kidney grafting
was performed; however, it partially failed due to recurrent
nephrolithiasis. Renal function in the patient deteriorated soon
after renal transplantation, and primary hyperoxaluria was
diagnosed ~4 weeks post-surgery when crystal deposition in the
renal tubules was demonstrated. This suggests that calcium oxalate
deposition occurs early after renal transplant, jeopardizing renal
function recovery and indicating that renal transplant may not be a
viable treatment option for type 2 patients.
In conclusion, although primary hyperoxaluria type 2
is rare, it should be considered in patients with recurrent
episodes of nephrolithiasis, particularly those who are
unresponsive to conventional therapies. Blood oxalate and stone
components should be examined in such patients. Combined
liver-kidney transplant may be required, as kidney transplant alone
is unlikely to be successful.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of China (grant no. 81501381), Science and
Technology Development Project of Jilin Province (grant no.
20160101100JC), Youth Fund of Health and Family Planning of Jilin
Province (grant no. 2015Q004).
Availability of data and materials
The datasets during and/or analyzed during the
current study available from the corresponding author on reasonable
request.
Authors' contributions
SL, BG and HZ mainly participated in the literature
search, study design, writing and critical revision. GW, WW, XL,
SW, JY and YF mainly participated in data collection, data analysis
and data interpretation. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was carried out in accordance with the
approved guidelines by the Ethics Committee of First Hospital of
Jilin University and the patients provided their signed and
informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hoppe B, Beck BB and Milliner DS: The
primary hyperoxalurias. Kidney Int. 75:1264–1271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barratt TM, Danpure Hyperoxaluria CJ,
Holliday MA, et al: Paediatric Nephrology. 3rd. Published Williams
and Wilkins; Baltimore: pp. 557–572. 1994
|
|
3
|
Wuhl E, van Stralen KJ, Wanner C, Ariceta
G, Heaf JG, Bjerre AK, Palsson R, Duneau G, Hoitsma AJ, Ravani P,
et al: Renal replacement therapy for rare diseases affecting the
kidney: An analysis of the ERA-EDTA registry. Nephrol Dial
Transplant. 29 Suppl 4:Siv1–Siv8. 2014. View Article : Google Scholar
|
|
4
|
Hulton SA: The primary hyperoxalurias: A
practical approach to diagnosis and treatment. Int J Surg.
36:649–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Danpure CJ and Rumsby G: Molecular
aetiology of primary hyperoxaluria and its implications for
clinical management. Expert Rev Mol Med. 6:1–16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marangella M, Petrarulo M, Cosseddu D,
Vitale C, Cadario A, Barbos MP, Gurioli L and Linari F: Detection
of primary hyperoxaluria type 2 (L-glyceric aciduria) in patients
with maintained renal function or end-stage renal failure. Nephrol
Dial Transplant. 10:1381–1385. 1995.PubMed/NCBI
|
|
7
|
Tang X, Bergstralh EJ, Mehta RA, Vrtiska
TJ, Milliner DS and Lieske JC: Nephrocalcinosis is a risk factor
for kidney failure in primary hyperoxaluria. Kidney Int.
87:623–631. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hicks NR, Cranston DW and Charlton CA:
Fifteen-year follow-up of hyperoxaluria type II. N Engl J Med.
309:7961983. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martin-Higueras C, Torres A and Salido E:
Molecular therapy of primary hyperoxaluria. J Inherit Metab Dis.
40:481–489. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rumsby G and Hulton SA: Primary
hyperoxaluria type 2 = Gene Reviews®. Adam MP, Ardinger
HH, Pagon RA and Wallace SE: University of Washington; Seattle, WA:
1993
|
|
11
|
Williams EL, Acquaviva C, Amoroso A,
Chevalier F, Coulter-Mackie M, Monico CG, Giachino D, Owen T,
Robbiano A, Salido E, et al: Primary hyperoxaluria type 1: Update
and additional mutation analysis of the AGXT gene. Hum Mutat.
30:910–917. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cochat P: Primary hyperoxaluria type 1.
Kidney Int. 55:2533–2547. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harambat J, Fargue S, Bacchetta J,
Acquaviva C and Cochat P: Primary hyperoxaluria. Int J Nephrol.
2011:8645802011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Topaloğlu R, Bakkaloğlu A, Saatçi U and
Beşbaş N: Early onset of stone diseases and primary hyperoxaluria.
Int Urol Nephrol. 22:223–226. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kasidas GP: Plasma and urine measurements
for monitoring of treatment in the primary hyperoxaluric patient.
Nephrol Dial Transplant. 10 Suppl 8:S8–S10. 1995. View Article : Google Scholar
|
|
16
|
Danpure CJ and Jennings PR: Further
studies on the activity and subcellular distribution of
alanine:glyoxylate aminotransferase in the livers of patients with
primary hyperoxaluria type 1. Clin Sci (Lond). 75:315–322. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Milosevic D, Rinat C, Batinic D and
Frishberg Y: Genetic analysis-a diagnostic tool for primary
hyperoxaluria type I. Pediatr Nephrol. 17:896–898. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rumsby G: An overview of the role of
genotyping in the diagnosis of the primary hyperoxalurias. Urol
Res. 33:318–320. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Naderi G, Latif A, Tabassomi F and
Esfahani ST: Failure of isolated kidney transplantation in a
pediatric patient with primary hyperoxaluria type 2. Pediatr
Transplant. 18:E69–E73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moray G, Tezcaner T, Özçay F, Baskın E,
Akdur A, Kırnap M, Yıldırım S, Arslan G and Haberal M: Liver and
kidney transplant in primary hyperoxaluria: A single center
experience. Exp Clin Transplant. 13 Suppl 1:S145–S147. 2015.
|
|
21
|
Bergstralh EJ, Monico CG, Lieske JC,
Herges RM, Langman CB, Hoppe B and Milliner DS; IPHR Investigators,
: Transplantation outcomes in primary hyperoxaluria. Am J
Transplant. 10:2493–2501. 2010. View Article : Google Scholar : PubMed/NCBI
|