Introduction
Chronic kidney disease (CKD) is a worldwide health
concern with high morbidity and mortality (1). Tubulointerstitial fibrosis is a major
pathological characteristic of CKD (2) and renal tubular epithelial cell (RTEC)
damage induces interstitial fibrosis (3). Therefore, elucidating the mechanism
responsible for RTEC injury is critical for developing a treatment
to prevent fibrosis and interrupt the progression of CKD.
It has previously been reported that macroautophagy
(autophagy) is associated with RTEC damage (4). Autophagy is a highly regulated
lysosomal protein degradation process in which damaged organelles
and long-lived proteins are removed in order to maintain
intracellular homeostasis and cellular integrity (5). During autophagy activation, the
biosynthesis of microtubule-associated protein 1 light chain 3B-II
(LC3-II) (6) and B-cell lymphoma-2
(Bcl-2)-interacting myosin-like coiled-coil protein (Beclin1)
(7) increases. These proteins are
associated with the extent of autophagosome formation (8). p62 is known to serve a role in linking
polyubiquitinated protein aggregates in the autophagic machinery
and upregulated protein expression indicates inhibited autophagy
(9). Yang et al (10) reported that the nephrotoxic drug
cisplatin induced autophagic activation to protect RTEC from
apoptosis. By contrast, Suzuki et al (11) demonstrated that, in renal
ischemia/reperfusion injury, increased autophagic activity resulted
in RTEC death. As such, it is not known whether autophagy has a
protective or detrimental effect on cell survival.
In the pathogenesis of CKD, autophagy serves a role
in RTEC injury when exposed to harmful conditions, including
excessive proteinuria (12) and high
glucose (13). Advanced oxidation
protein products (AOPPs), which are primarily formed by the
reaction of plasma albumin with chlorinated oxidants (14), were revealed to be elevated in the
plasma of patients with CKD and the level of AOPPs is positively
associated with the deterioration of renal function in CKD
(15). Zhou et al (16) demonstrated that AOPPs induced
glomerular podocyte apoptosis and albuminuria in normal rats and Li
et al (17) suggested that
AOPPs induced renal fibrosis in the remnant kidneys of 5/6
nephrectomy rats. A previous study by our group demonstrated that
AOPPs induced RTEC injury, including hypertrophy and
epithelial-to-mesenchymal transition (EMT) (18). However, the effects of AOPPs on RTEC
autophagy are yet to be investigated.
Notably, autophagy activation is modulated by
cross-linked signaling pathways (19). The kinase mammalian target of
rapamycin (mTOR) is the key modulator of autophagy and acts as a
‘gatekeeper’ of various upstream signaling pathways (20). In recent years, researchers have
taken great interest in the phosphoinositide 3-kinase
(PI3K)/protein kinase B (AKT)/mTOR pathway, which is a crucial
negative modulator of autophagy (21). Furthermore, a number of studies have
demonstrated that activation of this signaling pathway serves a
crucial role in the pathogenesis of CKD (22,23).
Therefore, it is reasonable to hypothesize that the PI3K/AKT/mTOR
pathway may be associated with the effects of AOPPs on RTEC
autophagy.
The aim of the present study was to examine the
effect of AOPPs on autophagy and the probable modulatory pathway in
cultured HK-2 cells in vitro in order to elucidate the
mechanism of AOPP-induced RTEC injury.
Materials and methods
AOPPs-bovine serum albumin (BSA)
preparation and content determination
AOPPs were prepared as previously described
(24). BSA was obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and hypochlorous
acid (HOCl) was purchased from Fluke Switzerland GmbH (Basserdorf,
Switzerland). BSA solution (100 mg/ml) was combined with HOCl (200
mmol/l) at a molar ratio of 1:140 for 30 min at room temperature in
the dark and in the absence of free amino acid/carbohydrates/lipids
to exclude the formation of advance glycation end product-like
structures. Prepared samples were dialyzed in 4°C PBS to remove
free HOCl for 24 h. BSA was dissolved in PBS alone as control. All
samples were passed through a Detoxi-Gel column (Pierce; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) to remove contaminating
endotoxins. The endotoxin levels were measured using the Amebocyte
lysate assay kit (Sigma Aldrich; Merck KGaA) and were demonstrated
to be <0.025 EU/ml. The AOPP content was determined by measuring
absorbance at 340 nm in an acidic condition after mixing 200 µl of
prepared sample or chloramines-T with 20 µl of acetic acid in a
96-well plate. The sample was calibrated using chloramines-T in the
presence of potassium iodide (14).
Culture of HK-2
Immortalized HK-2 cells were purchased from ATCC
(Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's
medium, nutrient mixture F12 (DMEM/F12; Hyclone; GE Healthcare,
Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine
serum (FBS; Gibco, Thermo Fisher Scientific, Inc.) and an
antibiotic mixture of penicillin (100 U/ml) and streptomycin (100
µg/ml) at 37°C in an atmosphere containing 5% CO2. Cells
at 80% confluence cells from passages 2 to 5 were used. In the
LY294002 (CST Biological Reagents Co., Ltd.), rapamycin
(Sigma-Aldrich; Merck KGaA) and chloroquine (Sigma Aldrich; Merck
KGaA) blocking experiments, cells were pretreated with LY294002 (10
µM), rapamycin (1 µM) or chloroquine (1 mM) for 1 h at room
temperature and subsequently treated with AOPPs for 24 h at room
temperature.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. cDNA was synthesized using total RNA (1
µg) with random primers and the MMLV reverse transcriptase First
Strand kit at 37°C for 15 min, followed by 85°C for 5 sec and
storage at 4°C (Invitrogen; Thermo Fisher Scientific, Inc.). qPCR
was performed using the SYBR®Premix Ex
Taq™kit (Takara Bio, Inc., Otsu, Japan). The
thermocycling conditions were as follows: 95°C for 10 min, followed
by 40 cycles of 95°C for 10 sec and 60°C for 30 sec. The following
primers were used: GAPDH forward, 5′-CGGAGTCAACGGATTTGGTCGTAT-3′
and reverse, 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′; kidney injury molecule
1 (KIM-1) forward, 5′-GACAACGAGCATTCCAACAA-3′ and reverse,
5′-GCTGAGGTGAAGATGGTGAAG-3′; neutrophil gelatinase-associated
lipocalin (NGAL) forward, 5′-ACAAAGACCCGCAAAAGATG-3′ and reverse,
5′-GCAACCTGGAACAAAAGTCC-3′. All data was normalized using the
internal control GAPDH. Fold change were quantified using the
2−∆∆Cq method (25).
Western blotting
Total protein was extracted from cells using
pre-cooled radioimmunoprecipitation assay lysis buffer containing
cocktail protease inhibitors (Biotool; Stratech Scientific, Ltd.,
Newmarket, UK). Protein concentrations were determined using the
Micro Bicinchoninic Acid Assay kit (CoWin Biosciences, Beijing,
China). According to the expression abundance and molecular weight
of the proteins, 40 µg of LC3B and p62 were separated using 12%
SDS-PAGE and 20 µg of the remaining proteins were separated using
8% SDS-PAGE. Proteins were then transferred onto polyvinylidene
fluoride membranes. Membranes were blocked in 5% non-fat milk at
room temperature for 1–3 h, followed by incubation with primary
antibodies for 2 h at room temperature and additional incubation at
4°C overnight. The primary antibodies included the following:
Anti-LC3B (cat. no. 2775 1:1,000), anti-phospho-AKT (Ser 473; cat.
no. 4060; 1:1,000), anti-Beclin1 (cat. no. 3738; 1:1,000), anti-AKT
(cat. no. 9272; 1:2,000), anti-p62 (cat. no. 8025; 1:2,000; all CST
Biological Reagents Co., Ltd.), anti-p85 PI3K (cat. no. WL01169;
1:500; Wanleibio Co., Ltd., Shanghai, China), anti-phospho-mTOR
(Ser 2448; cat. no. sc-293133; 1:500), anti-mTOR (cat. no. sc8319;
1:500; both Santa Cruz Biotechnology, Inc., Dallas, TX, USA), and
anti-GAPDH (cat. no. 20301707-1; 1:5,000; Bioworld, Dublin, OH
USA). The membranes were washed three times with TBS containing
Tween-20 (TBST) for 10 min and subsequently incubated with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (cat. no. bs-0295M-HRP; 1:10,000; BIOSS, Beijing, China)
at room temperature for 1 h. Membranes were washed three times with
TBST for 10 min and proteins were detected using an enhanced
chemiluminescence system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Semi-quantitative analysis was performed using ImageJ
software (version 1.48u; National Institutes of Health, Bethesda,
MD, USA). GAPDH was used as an internal control.
ELISA assay
HK-2 cells were cultured and treated with 200 µg/ml
AOPPs at room temperature for 24 h in 96-well plates. The collected
cell culture medium was centrifugated at a speed of 95 × g for 5
min at room temperature to obtain the supernatant and discard the
cell debris. All reagents and samples were brought to room
temperature prior to use. Monoclonal antibodies [cat. no.
70-EK11181; 1:100; Hangzhou MultiSciences (Lianke) Biotech Co.,
Ltd., Hangzhou, China] specific to human KIM-1 or NGAL were
pre-coated onto microplates with carbonate buffer of (pH 9.6)
overnight at 4°C. A total of 100 µl of standards (concentrations
ranging from 1–2,000 pg/ml) or samples were added to each well in
triplicate, followed by 50 µl of diluted KIM-1 and NGAL detection
antibodies [cat. no. 70-EK11181; 1:100; Hangzhou MultiSciences
(Lianke) Biotech Co., Ltd.]. Plates were incubated at room
temperature for 2 h with gentle agitation. Wells were washed six
times and 100 µl of diluted streptavidin-horseradish
peroxidase-conjugated secondary antibody (cat. no. 70-EK11181;
1:100; Multi Science, Hangzhou, China) was added and incubated at
room temperature for 45 min with gentle agitation. Plates were
washed six times and 100 µl of substrate solution [cat. no.
70-EK11181; Hangzhou MultiSciences (Lianke) Biotech Co., Ltd.] was
added at room temperature for 30 min in the dark. Finally, the
absorbance was read at 450 and 570 nm using a microplate
reader.
Immunofluorescence
HK-2 cells were seeded at density of
1,000/cm2 in a laser confocal dish. Following treatment,
the supernatant was discarded and cells were washed three times
with PBS for 5 min. Cells were subsequently fixed with 4%
paraformaldehyde at room temperature for 15 min. Following washing
with PBS, cells were permeabilized with 0.3% Triton X-100 for 15
min and incubated in a blocking buffer containing 5% BSA for 30 min
at room temperature. Cells were incubated with anti-LC3B antibody
(ca. no. 2775; 1:100; CST Biological Reagents Co., Ltd.) for 1 h at
room temperature and again overnight at 4°C. Secondary antibodies
labeled with fluorescein (cat. no. SA00006-6; Alexa Fluor 488;
1:1,000; Proteintech Group, Inc., Chicago, IL, USA) were added and
incubated for 1 h at 37°C in the dark. Cells were subsequently
incubated with 0.1% DAPI for 10 min at room temperature and washed
with PBS. The cells were observed using a laser confocal
fluorescent microscope at a magnification of ×100. The detection of
punctuated LC3B staining in the diffuse staining indicated the
quantity of autophagosomes. LC3B-stained dots were counted in
individual HK-2 cells and the mean of ≥30 cells was calculated.
Transmission electron microscopy
HK-2 cells were harvested gently using trypsin-EDTA
(Gibco; Thermo Fisher Scientific, Inc.) following 12 h of culture.
Cells were centrifuged at a speed of 95 × g for 5 min at room
temperature and the sediment cells were collected, washed twice
with cold PBS and fixed in 2.5% glutaraldehyde for 2 h at 4°C.
Cells were conventionally dehydrated, embedded, sliced into 60 nm
sections and stained with uranyl acetate for 15 min at room
temperature, followed by lead citrate for 15 min at room
temperature. Autophagosome (AP) and autolysosome (AL) formation was
observed using transmission electron microscopy (TEM) at a
magnification of ×10,000 or ×20,000. During the TEM study, images
of 10 random fields were captured in a grid (top left, middle left,
bottom left, center, top right, middle right and bottom right) and
the number of APs, ALs and cells per field was counted.
Statistical analysis
All experiments were performed in triplicate. Data
are presented as the mean ± standard deviation. Differences between
groups were assessed using one-way analysis of variance followed by
a post hoc Tukey's test. When comparing 2 groups, Fisher's Least
Significant Difference method was used. When the assumption of
equal variance did not hold, the Dunnett's T3 method was used.
Statistical analyses were performed using SPSS 20.0 (IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
AOPPs inhibit autophagic activity in
HK-2 cells
To investigate the effects of AOPPs on the autophagy
of RTECs, western blotting was performed to examine the expression
of LC3B, Beclin1 and p62 proteins (Fig.
1A and B). AOPPs treatment significantly upregulated the
expression of p62, whereas the expression of Beclin1 and conversion
of LC3-I to LC3-II were significantly downregulated compared with
the control group. However, no significant differences were
observed between the control and unmodified BSA groups. The optimal
concentration and duration of treatment were determined to be 200
µg/ml and 12 h, respectively (Fig. 1C
and D). HK-2 cells were therefore treated with control medium
or AOPPs (200 µg/ml) for 12 h in subsequent experiments.
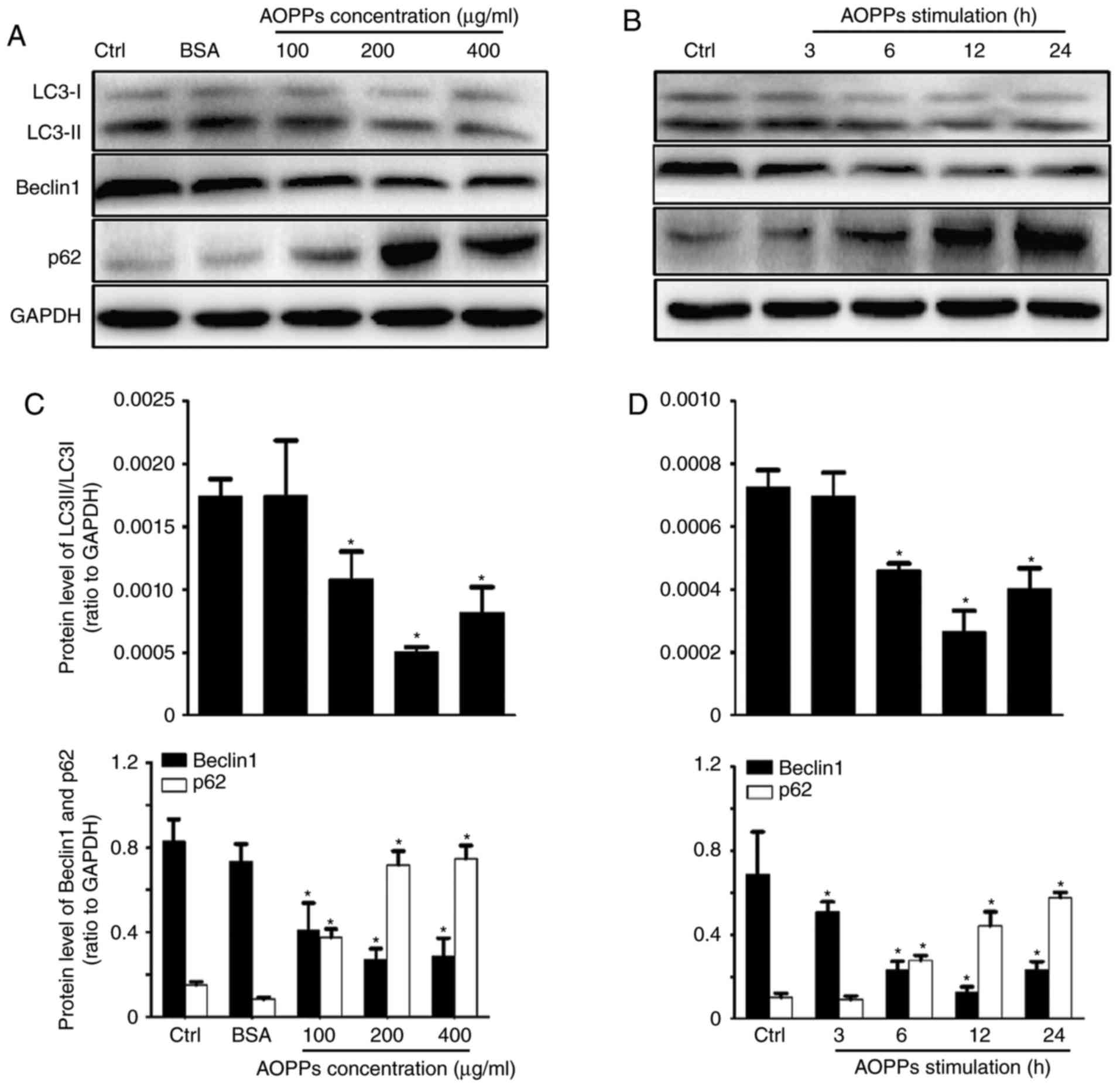 | Figure 1.AOPPs inhibit autophagic activity in
HK-2 cells. HK-2 cells were treated with (A) vehicle control,
unmodified BSA (200 µg/ml) and BSA + AOPPs for 12 h, or (B) 200
µg/ml of AOPPs for 3, 6, 12 or 24 h and levels of LC3-I, LC3-II,
Beclin1 and p62 were assessed using western blotting. (C and D)
Quantified results of western blotting. Data are expressed as the
mean ± standard deviation of three independent experiments;
*P<0.05 vs. Ctrl. AOPPs, advanced oxidation protein products;
BSA, bovine serum albumin; LC3, microtubule-associated proteins 1
light chain 3B; Beclin1, B-cell lymphoma-2-interacting myosin-like
coiled-coil protein; Ctrl, control. |
Autophagic activity of the HK-2 cells was assessed
using immunofluorescence technology and TEM (Fig. 2). Immunofluorescent staining revealed
that LC3B-positive staining was significantly decreased in the
AOPP-treated cells compared with the control group (Fig. 2A and C). Similarly, the TEM results
demonstrated a decrease in the number of typical APs, which are
recognized as double-membrane vacuoles engulfing cytoplasmic
structures and ALs, characterized as single-membrane vacuole
structures containing high-density materials, in AOPP-treated cells
compared with the control group (Fig. 2B
and D). Collectively, these results suggest that AOPPs inhibit
the activation of autophagy in HK-2 cells.
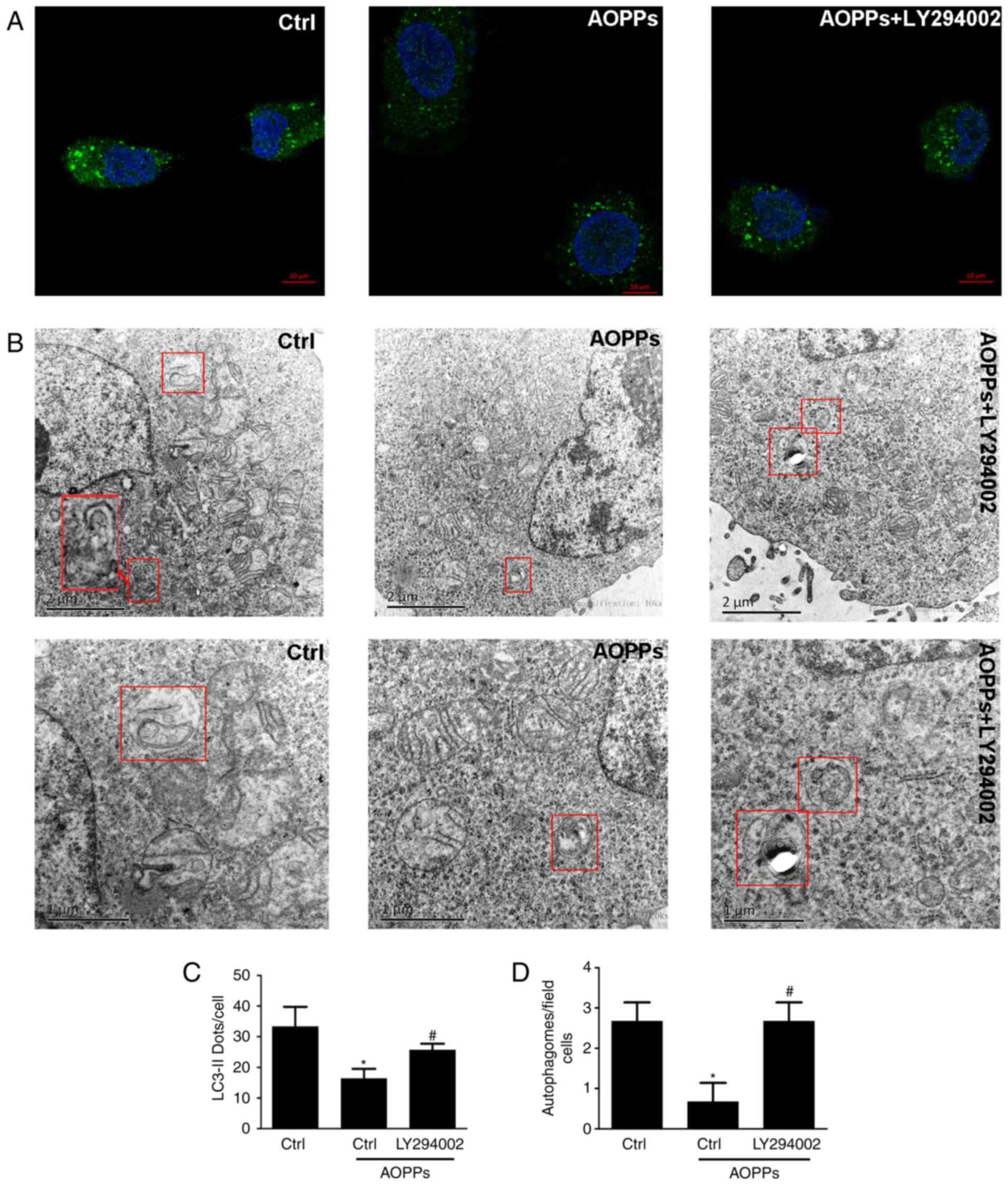 | Figure 2.Immunofluorescent staining and TEM
observation of autophagy. HK-2 cells were treated with the vehicle
control or 200 µg/ml AOPPs for 12 h in the absence or presence of
LY294002 (10 µM). (A) Indirect immunofluorescent staining revealed
that AOPP treatment decreased positive LC3B staining, whereas
LY294002 addition reversed this effect. Magnification, ×100. Scale
bar, 10 µm. (B) TEM visualization revealed that AOPP treatment
inhibited autophagic vacuole formation, whereas LY294002 treatment
improved it. Top row, magnification, ×10,000 and scale bar, 2 µm;
bottom row, magnification, ×20,000 and scale bar, 1 µm (C)
LC3B-positive cells were counted in individual HK-2 cells and the
mean of ≥30 cells was calculated. (D) Images of 10 random TEM
fields were captured in a grid (top left, middle left, bottom left,
center, top right, middle right and bottom right) and the number of
autophagosomes, autolysosomes and cells were counted. Data are
expressed as the mean ± standard deviation of three independent
experiments; *P<0.05 vs. control, #P<0.05 vs.
AOPP-treatment group. TEM, transmission electron microscopy; AOPPs,
advanced oxidation protein products; LC3B, microtubule-associated
proteins 1 light chain 3B; Ctrl, control. |
PI3K/AKT/mTOR signaling pathway
activation mediates AOPP-inhibited autophagy
Next, it was assessed whether the PI3K/AKT/mTOR
signaling pathway is associated with the AOPP-induced inhibition of
HK-2 autophagy. The expression of p85-PI3K, p-AKT (Ser473), AKT,
p-mTOR (Ser2448) and mTOR proteins in cells was measured following
exposure to AOPPs (Fig. 3).
Treatment with AOPPs induced the phosphorylation of PI3K, AKT and
mTOR. Specifically, at 6 h post-treatment, the significant
phosphorylation of PI3K, AKT and mTOR was observed and this
increase peaked at 12 h post-treatment (Fig. 3D and F). Furthermore, the optimal
concentration of AOPPs was determined to be 200 µg/ml (Fig. 3C and E). No significant differences
were observed between control and unmodified BSA-treated cells.
Collectively, these results suggest that AOPPs trigger the
PI3K/AKT/mTOR signaling pathway.
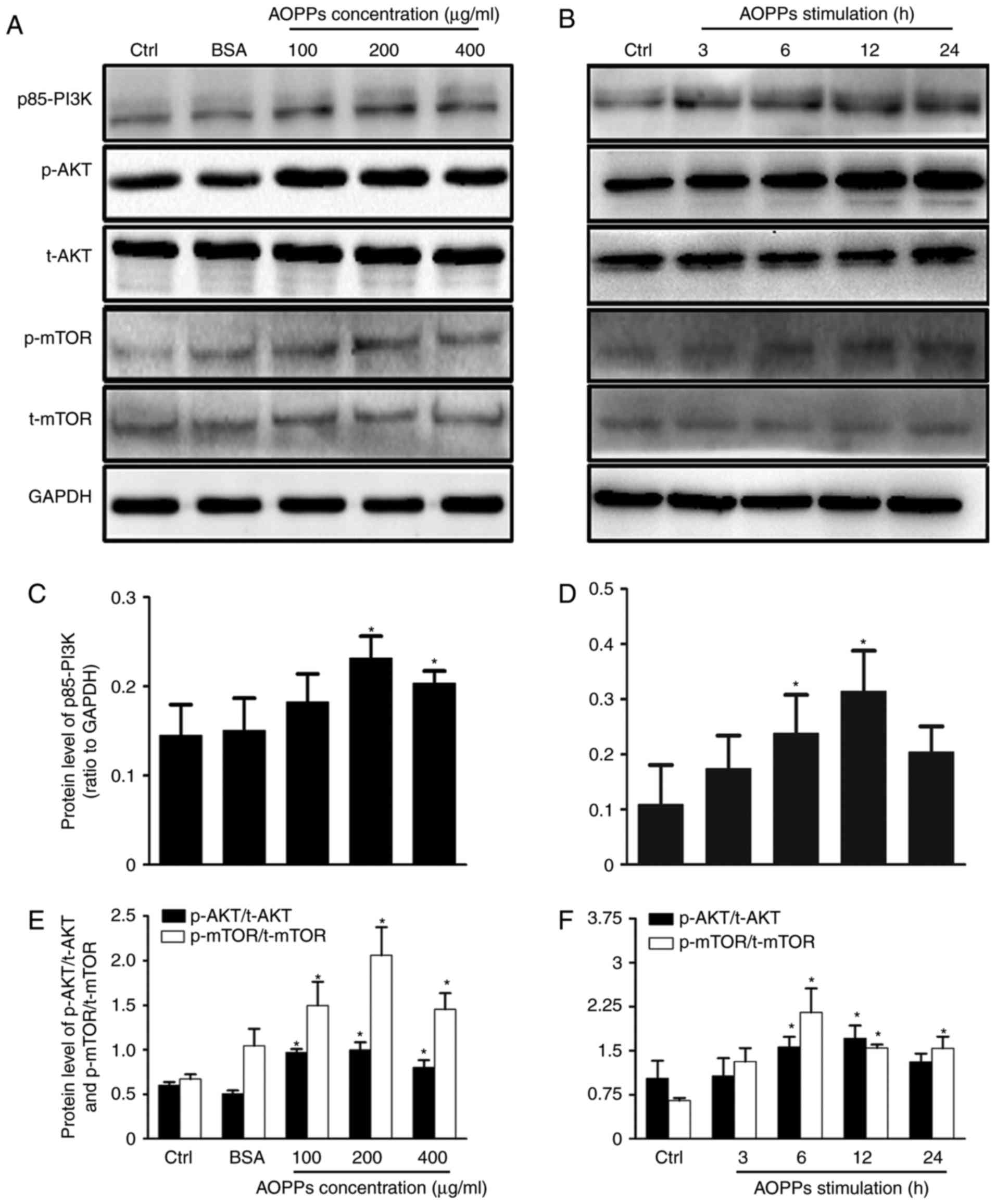 | Figure 3.AOPPs activate the PI3K/AKT/mTOR
signaling pathway. HK-2 cells were treated with (A) vehicle,
unmodified BSA (200 µg/ml) and 100, 200 or 400 µg/ml AOPPs for 12
h, or (B) 200 µg/ml of AOPPs for 3, 6, 12 or 24 h and the
phosphorylation of PI3K, AKT and mTOR was assessed using western
blotting (C-F). Western blotting results were quantified using
densitometry. Data are presented as the mean ± standard deviation
of three independent experiments; *P<0.05 vs. Ctrl. AOPPs,
advanced oxidation protein products; PI3K, phosphoinositide
3-kinase; AKT, protein kinase B; mTOR, mammalian target of
rapamycin; BSA, bovine serum albumin; Ctrl, control; p,
phosphorylated; t, total. |
Cells were subsequently treated with AOPPs for 12 h
in the presence or absence of LY294002 (10 µM), a PI3K inhibitor
that blocks the PI3K/AKT/mTOR pathway (Fig. 4). The AOPP-induced downregulation of
Beclin1 and LC3-II expression, as well as the AOPP-induced
upregulation of p62 was significantly reversed by treatment with
LY294002 (Fig. 4D). Furthermore,
LC3B-positive staining was significantly increased in cells treated
with LY294002 compared with those treated with AOPPs alone
(Fig. 2A and C). Electron microscopy
analysis demonstrated that LY294002 also partially ameliorated the
AOPP-induced decrease in APs and ALs (Fig. 2B and D). These results suggest that
the PI3K/AKT/mTOR signaling pathway mediates the AOPP-induced
inhibition of autophagic activity in HK-2 cells.
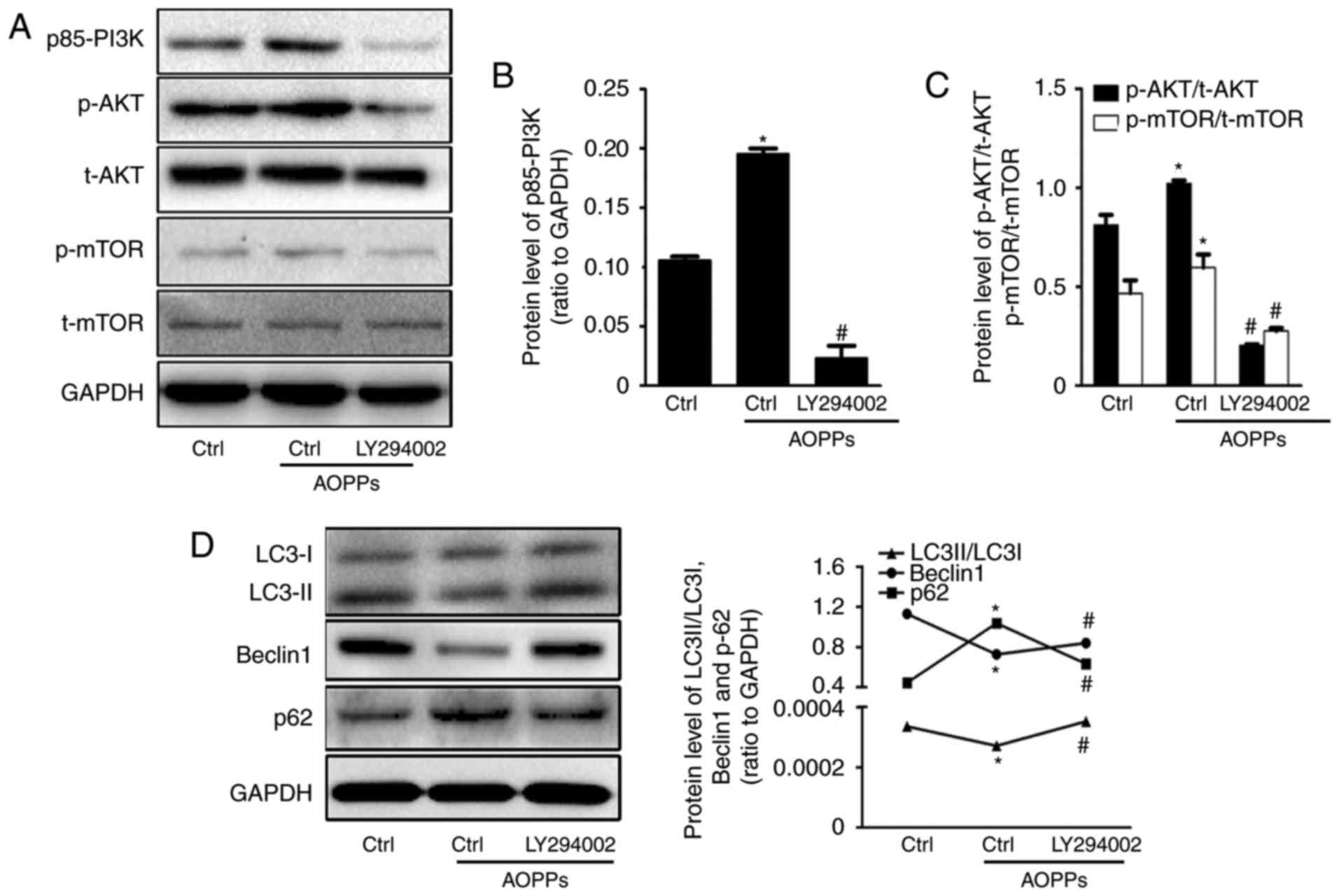 | Figure 4.AOPPs inhibit autophagy via the
PI3K/AKT/mTOR signaling pathway. (A) HK-2 cells were treated with
the vehicle control or 200 µg/ml AOPPs for 12 h in the absence or
presence of 10 µM LY294002 and the expression of (B) PI3K, (C) AKT
and mTOR was quantified. The PI3K/AKT/mTOR signaling pathway was
significantly blocked by LY294002. (D) Western blotting revealed
that the AOPP-induced decrease in LC3-I to LC3-II conversion,
suppression of Beclin1 and overexpression of p62 were partly
reversed by treatment with LY294002. Data are presented as the mean
± standard deviation of three independent experiments. *P<0.05
vs. Ctrl, #P<0.05 vs. AOPP-treatment group. AOPPs,
advanced oxidation protein products; PI3K, phosphoinositide
3-kinase; AKT, protein kinase B; mTOR, mammalian target of
rapamycin; LC3, microtubule-associated proteins 1 light chain 3B;
Beclin1, B-cell lymphoma-2-interacting myosin-like coiled-coil
protein; Ctrl, control; p, phosphorylated; t, total. |
Autophagy inhibition mediates
AOPP-induced injury to RTECs
To explore the effect of autophagy inhibition on
AOPP-induced RTEC injury, RTEC injury was assessed by measuring the
expression of two novel renal tubular injury markers, KIM-1 and
NGAL, using RT-qPCR and ELISA. KIM-1 is overexpressed on the
surface of proximal tubular cells and NGAL is produced
predominantly by neutrophils and in part by proximal tubular cells;
both are produced in response to harmful stimuli and act as
indicators of RTEC injury (26).
Compared with the control group, the expression of KIM-1 and NGAL
mRNA and protein was significantly upregulated in cells treated
with AOPPs (Fig. 5). No significant
difference was observed in unmodified BSA-treated cells, indicating
that the overexpression of KIM-1 and NGAL was associated with
advanced oxidation of BSA. Additionally, a significant increase in
KIM-1 and NGAL levels was observed in the cell supernatant at 24 h
post-treatment, which is consistent with the excretion
characteristics of the two proteins (26).
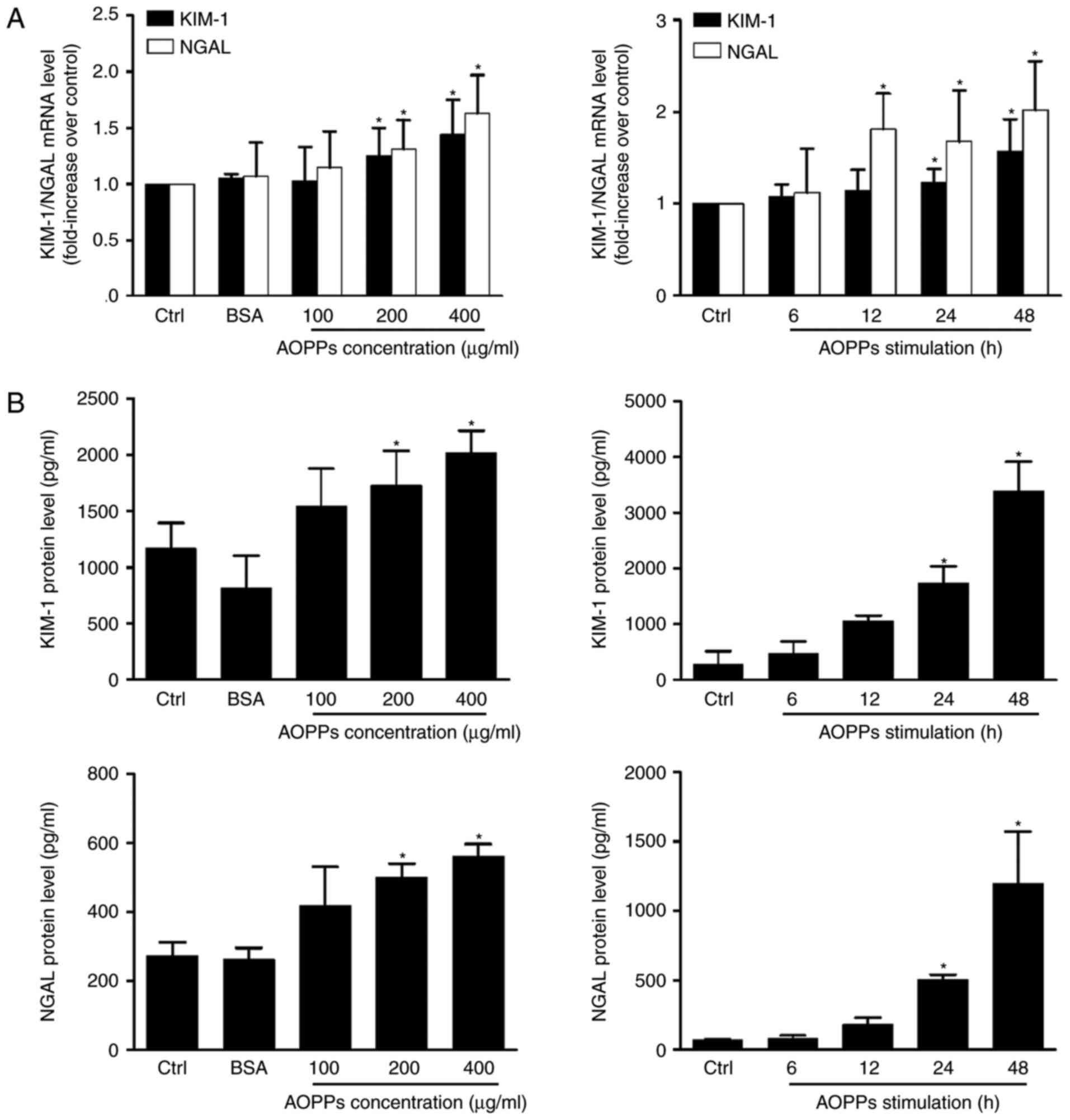 | Figure 5.AOPPs induce HK-2 cell injury. HK-2
cells were cultured in control medium, native BSA (200 µg/ml) and
100, 200 and 400 µg/ml AOPPs for 24 h, or 200 µg/ml of AOPPs for 6,
12, 24 and 48 h. (A) Reverse transcription-quantitative polymerase
chain reaction demonstrated that AOPP treatment increases the
expression of KIM-1 and NGAL mRNA. (B) ELISA assays revealed that
AOPP treatment ≥200 µg/ml induced a significant increase in KIM-1
and NGAL expression following 24 h. Data is expressed as the mean ±
standard deviation of three independent experiments. *P<0.05 vs.
Ctrl. AOPPs, advanced oxidation protein products; BSA, bovine serum
albumin; KIM-1, kidney injury molecular; NGAL, neutrophil
gelatinase-associated lipocalin; PI3K, phosphoinositide 3-kinase;
ctrl, control. |
Cells were pretreated with rapamycin, an autophagy
inducer, or chloroquine, an autophagy inhibitor, for 1 h prior to
treatment with AOPPs for 24 h. The results of western blotting
revealed that rapamycin (1 µM) significantly increased autophagy,
whereas chloroquine (1 mM) inhibited it (Fig. 6A). RT-qPCR (Fig. 6B) and ELISA (Fig. 6C and D) detection of KIM-1 and NGAL
revealed a partial but significant decrease in the
rapamycin-treatment group as well as an increase in the
chloroquine-treatment group compared with AOPP treatment alone.
These data suggest that autophagy inhibition serves a role in
AOPP-induced HK-2 cell injury.
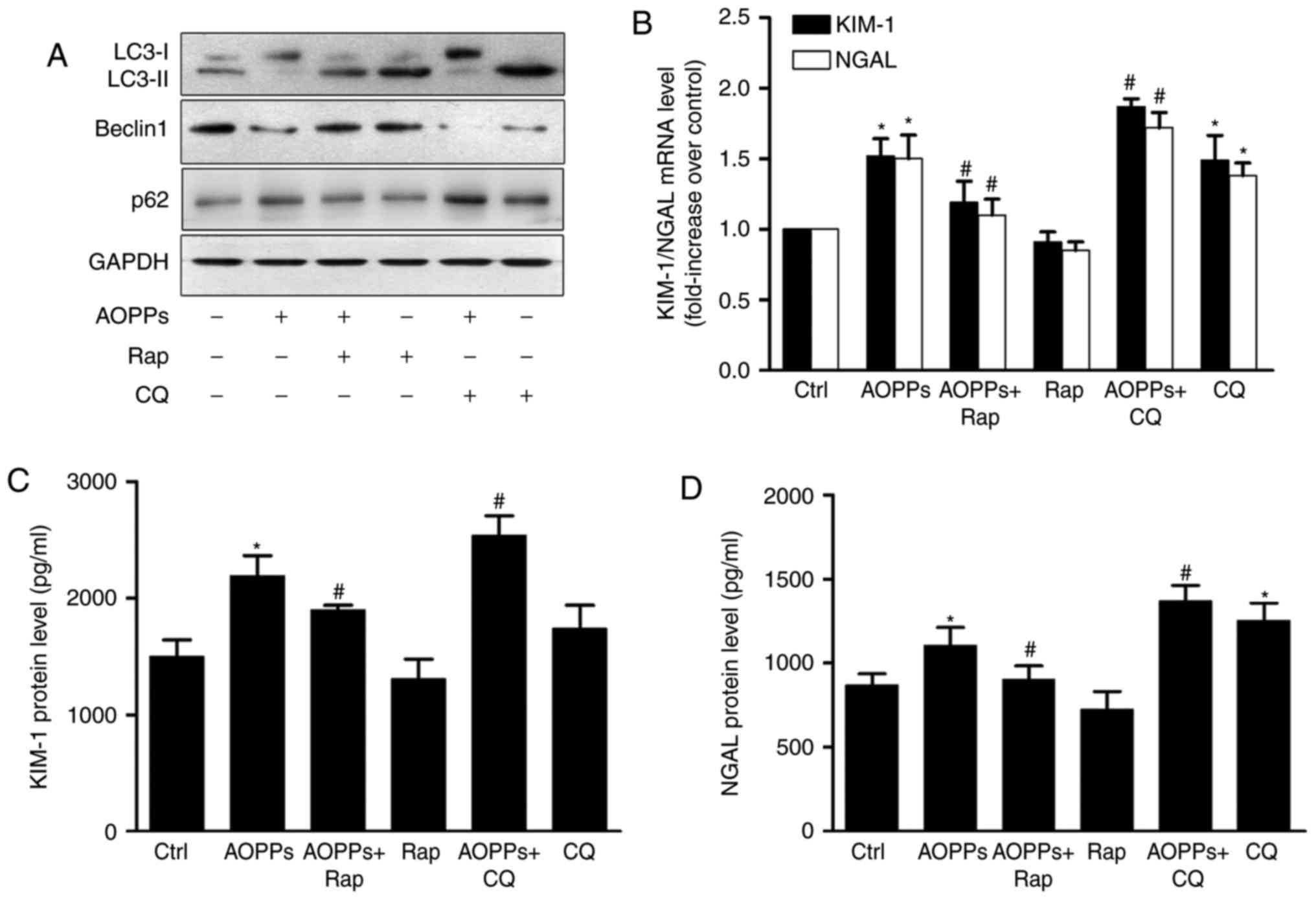 | Figure 6.Autophagy inhibition mediates
AOPP-induced HK-2 injury. HK-2 cells were treated with vehicle
control or 200 µg/ml AOPPs for 24 h, in the absence or presence of
rap (1 µM) or CQ (1 mM). (A) Western blotting demonstrated that rap
increased autophagy in AOPP-treated HK-2 cells, whereas CQ
decreased it. (B) Reverse transcription-polymerase chain reaction
revealed that the AOPP-induced over-expression of KIM-1 and NGAL
mRNA was reversed by rap and further aggravated by CQ. ELISA assays
demonstrated that the AOPP-induced over-expression of (C) KIM-1 and
(D) NGAL protein was reversed by rap or further aggravated by CQ.
Data are presented as the mean ± standard deviation of three
independent experiments. *P<0.05 vs. Ctrl, #P<0.05
vs. AOPP-treatment group. AOPP, advanced oxidation protein product;
rap, rapamycin; CQ, chloroquine; KIM-1, kidney injury molecular;
NGAL, neutrophil gelatinase-associated lipocalin; PI3K,
phosphoinositide 3-kinase; Beclin1, B-cell lymphoma-2-interacting
myosin-like coiled-coil protein; LC3, microtubule-associated
proteins 1 light chain 3B;ctrl, control. |
Discussion
In the present study it was demonstrated that
autophagy inhibition mediates AOPP-induced HK-2 cell injury in CKD,
which is modulated by the PI3K/AKT/mTOR signaling pathway.
Autophagy is a dynamic process that comprises autophagosome
formation, lysosome fusion and turnover in the lysosome (5). Although the autophagosome is formed
during the initiation of autophagy, autophagic flux may be blocked
if the fusion and/or turnover processes fail (9,27). Liu
et al (12) demonstrated that
proteinuria exposure increased the expression of LC3-II and
decreased p62 levels, suggesting that proteinuria may induce
autophagy initiation as well as autophagosome degradation. Huang
et al (13) reported that
high glucose induced the overexpression of p62 in synchrony with
LC3-II upregulation, which indicates that high glucose inhibits the
turnover of autophagosomes. The results of the present study
indicate that the expression of p62 is upregulated when LC3-II and
Beclin1 expression, as well as autophagosome formation, are
downregulated in cells treated with AOPP. This suggests that AOPPs
inhibit the initiation of autophagy.
Autophagy activation is modulated by complex
upstream signaling pathways (28).
As AOPPs may inhibit the initiation of autophagy in RTECs, the
probable modulatory pathway was investigated. In agreement with
previous studies, which have reported that the PI3K/AKT/mTOR
signaling pathway serves a crucial role in the pathogenesis of CKD
(20,29), it was demonstrated that this
well-known negative modulatory pathway mediated AOPP-inhibited
autophagy in RTECs.
A number of studies have reported that autophagy
serves a role in CKD (30,31). Yamahara et al (32) reported that obesity-mediated
autophagy insufficiency exacerbated proteinuria-induced
tubulointerstitial lesions, while Kitada et al (33) demonstrated that RTEC autophagy was
impaired in diabetic nephropathy and dietary restriction improved
autophagy to alleviate RTEC injury. AOPPs accumulation is a sign of
renal failure as well as an executor of renal deterioration
(14), which also induced an
increase in RTEC injury biomarkers KIM-1 and NGAL via inhibiting
autophagy in the present study. KIM-1 and NGAL have previously been
reported to be useful biomarkers for the assessment of renal
tubular injury (26). Liu et
al (12) demonstrated that these
biomarkers were significantly increased when HK-2 cells were
treated with urinary protein. However, Huang et al (34) reported that limited overexpression of
KIM-1 and NGAL was observed when cells were exposed to nephrotoxic
substances, including cisplatin. Therefore, only the KIM-1 and NGAL
alone are insufficient to determine how autophagy mediates HK-2
cells injury. Future studies should investigate the correlation
between autophagy and hypertrophy and EMT, as well as the injury
phenotype of RTEC, in order to further elucidate the effect of
autophagy on AOPP-induced RTEC injury. The findings presented here
were obtained from an in vitro study only; appropriate
animals models and primary cell models should be constructed to
determine the role of AOPPs in vivo. Finally, autophagy
induction is modulated by cross-linked upstream signaling pathways
and the PI3K/AKT/mTOR signaling pathway is highly regulated by
multiple mechanisms, so more experiments should be performed to
clarify the relevant pathways and the function of the PI3K/AKT/mTOR
signaling pathway in AOPP-treated cells.
In conclusion, the results of the present study
suggest that AOPPs inhibit autophagy via activation of the
PI3K/AKT/mTOR pathway in HK-2 cells. On the basis of these
findings, AOPP-induced autophagy inhibition appears to be
associated with RTEC injury. These findings suggest that targeting
autophagy may be an effective therapeutic strategy for inhibiting
CKD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation of China (grant no. 81202842).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ designed the study. XX conducted most of the
experiments and helped design the study. SS assisted with the
culture of cells and qPCR experiments. CZ and YL jointly helped
with the immunofluorescence experiment. TJ and WZ jointly helped
with the transmission electron microscopy. TG was responsible for
the data analysis. XL and XT were responsible for revising and
submitting the manuscript and figures, gave their final approval of
the version to be published, and agree to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the study. All authors
collaborated to interpret the results and develop the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors have no conflict of interest to
declare.
Glossary
Abbreviations
Abbreviations:
|
AOPPs
|
advanced oxidation protein
products
|
|
AKT
|
protein kinase B
|
|
Beclin1
|
B-cell lymphoma-2-interacting
myosin-like coiled-coil protein
|
|
KIM-1
|
kidney injury molecular
|
|
LC3B
|
microtubule-associated proteins 1
light chain 3B
|
|
mTOR
|
mammalian target of rapamycin
|
|
NGAL
|
neutrophil gelatinase-associated
lipocalin
|
|
PI3K
|
phosphoinositide 3-kinase
|
References
|
1
|
Couser WG, Remuzzi G, Mendis S and Tonelli
M: The contribution of chronic kidney disease to the global burden
of major noncommunicable diseases. Kidney Int. 80:1258–1270. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eddy AA: Progression in chronic kidney
disease. Adv Chronic Kidney Dis. 12:353–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grgic I, Campanholle G, Bijol V, Wang C,
Sabbisetti VS, Ichimura T, Humphreys BD and Bonventre JV: Targeted
proximal tubule injury triggers interstitial fibrosis and
glomerulosclerosis. Kidney Int. 82:172–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huber TB, Edelstein CL, Hartleben B, Inoki
K, Jiang M, Koya D, Kume S, Lieberthal W, Pallet N, Quiroga A, et
al: Emerging role of autophagy in kidney function, diseases and
aging. Autophagy. 8:1009–1031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bjorkoy G, Lamark T, Pankiv S, Øvervatn A,
Brech A and Johansen T: Monitoring autophagic degradation of
p62/SQSTM1. Methods Enzymol. 452:181–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang C, Kaushal V, Shah SV and Kaushal GP:
Autophagy is associated with apoptosis in cisplatin injury to renal
tubular epithelial cells. Am J Physiol Renal Physiol.
294:F777–F787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki C, Isaka Y, Takabatake Y, Tanaka H,
Koike M, Shibata M, Uchiyama Y, Takahara S and Imai E:
Participation of autophagy in renal ischemia/reperfusion injury.
Biochem Biophys Res Commun. 368:100–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu WJ, Luo MN, Tan J, Chen W, Huang LZ,
Yang C, Pan Q, Li B and Liu HF: Autophagy activation reduces renal
tubular injury induced by urinary proteins. Autophagy. 10:243–256.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang C, Lin MZ, Cheng D, Braet F, Pollock
CA and Chen XM: Thioredoxin-interacting protein mediates
dysfunction of tubular autophagy in diabetic kidneys through
inhibiting autophagic flux. Lab Invest. 94:309–320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Witko-Sarsat V, Friedlander M,
Capeillère-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers
P and Descamps-Latscha B: Advanced oxidation protein products as a
novel marker of oxidative stress in uremia. Kidney Int.
49:1304–1313. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kalousová M, Skrha J and Zima T: Advanced
glycation end-products and advanced oxidation protein products in
patients with diabetes mellitus. Physiol Res. 51:597–604.
2002.PubMed/NCBI
|
|
16
|
Zhou LL, Hou FF, Wang GB, Yang F, Xie D,
Wang YP and Tian JW: Accumulation of advanced oxidation protein
products induces podocyte apoptosis and deletion through
NADPH-dependent mechanisms. Kidney Int. 76:1148–1160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li HY, Hou FF, Zhang X, Chen PY, Liu SX,
Feng JX, Liu ZQ, Shan YX, Wang GB, Zhou ZM, et al: Advanced
oxidation protein products accelerate renal fibrosis in a remnant
kidney model. J Am Soc Nephrol. 18:528–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang X, Rong G, Bu Y, Zhang S, Zhang M,
Zhang J and Liang X: Advanced oxidation protein products induce
hypertrophy and epithelial-to-mesenchymal transition in human
proximal tubular cells through induction of endoplasmic reticulum
stress. Cell Physiol Biochem. 35:816–828. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang YP, Liang ZQ, Gu ZL and Qin ZH:
Molecular mechanism and regulation of autophagy. Acta Pharmacol
Sin. 26:1421–1434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lieberthal W and Levine JS: The role of
the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc
Nephrol. 20:2493–2502. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Satriano J and Sharma K: Autophagy and
metabolic changes in obesity-related chronic kidney disease.
Nephrol Dial Transplant. 28 Suppl 4:S29–S36. 2013. View Article : Google Scholar
|
|
23
|
Lieberthal W and Levine JS: Mammalian
target of rapamycin and the kidney. II. Pathophysiology and
therapeutic implications. Am J Physiol Renal Physiol.
303:F180–F191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Witko-Sarsat V, Friedlander M, Nguyen Khoa
T, Capeillère-Blandin C, Nguyen AT, Canteloup S, Dayer JM, Jungers
P, Drüeke T and Descamps-Latscha B: Advanced oxidation protein
products as novel mediators of inflammation and monocyte activation
in chronic renal failure. J Immunol. 161:2524–2532. 1998.PubMed/NCBI
|
|
25
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C-T method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Caplin B and Nitsch D: Urinary biomarkers
of tubular injury in chronic kidney disease. Kidney Int. 91:21–23.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mavroeidi V, Petrakis I, Stylianou K,
Katsarou T, Giannakakis K, Perakis K, Vardaki E, Stratigis S,
Ganotakis E, Papavasiliou S and Daphnis E: Losartan affects
glomerular AKT and mTOR phosphorylation in an experimental model of
type 1 diabetic nephropathy. J Histochem Cytochem. 61:433–443.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Z and Choi ME: Autophagy in kidney
health and disease. Antioxid Redox Signal. 20:519–537. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takabatake Y, Kimura T, Takahashi A and
Isaka Y: Autophagy and the kidney: Health and disease. Nephrol Dial
Transplant. 29:1639–1647. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamahara K, Kume S, Koya D, Tanaka Y,
Morita Y, Chin-Kanasaki M, Araki H, Isshiki K, Araki S, Haneda M,
et al: Obesity-mediated autophagy insufficiency exacerbates
proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol.
24:1769–1781. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kitada M, Takeda A, Nagai T, Ito H,
Kanasaki K and Koya D: Dietary restriction ameliorates diabetic
nephropathy through anti-inflammatory effects and regulation of the
autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa)
rats: A model of type 2 diabetes. Exp Diabetes Res.
2011:9081852011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang JX, Kaeslin G, Ranall MV, Blaskovich
MA, Becker B, Butler MS, Little MH, Lash LH and Cooper MA:
Evaluation of biomarkers for in vitro prediction of drug-induced
nephrotoxicity: Comparison of HK-2, immortalized human proximal
tubule epithelial and primary cultures of human proximal tubular
cells. Pharmacol Res Perspect. 3:e001482015. View Article : Google Scholar : PubMed/NCBI
|














