Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) represents
a worldwide health problem which accounts for 5% of all stroke
cases. Approximately 12% of the patients affected die before
medical care, 33% within 48 h and 50% within 30 days of aSAH.
Moreover, 50% of patients who survive the initial hemorrhage and
defeat vasospasm, frequently suffer from permanent disability
(1). Traditionally, research has
usually focused upon spasm of the large cerebral arteries, which is
considered as a determinant element of the complicated pathogenesis
of SAH. However, recent studies indicate that the intervention of
delayed vasospasm provides little help in the outcome of aSAH
patients (2). In these studies, it
was suggested that early brain injury (EBI), which is induced in
the first 72 h following initial bleeding, is usually linked to
poor outcome, such as brain edema, disruption of the blood-brain
barrier (BBB), and a sudden rise in intracranial pressure (1,3).
Accumulating evidence supports the early generation
of reactive oxygen species (ROS) and oxidative stress after SAH and
their association with EBI and delayed ischemic neurological
deficits (DINDs) (1,3). In clinical trials, a reduction in
antioxidant systems, and an increase in lipid peroxidation products
is found within 72 h of initial bleeding and correlates well with
poor clinical status and outcome (4). Therefore, antioxidants have
successfully been used to prevent oxidative stress and reduce EBI
in animals (5,6).
Nuclear factor erythroid-related factor 2 (Nrf2), a
basic leucine zipper redox-sensitive transcription factor,
regulates the expression of many antioxidant and phase II
detoxifying enzyme genes, such as heme oxygenase-1 (HO-1),
glutathione S-transferase (GST), NADP(H): Quinine oxidoreductase
(NQO), UDP-glucuronosyltransferase (UGT), and γ-glutamylcysteine
synthetase (γGCS) (7). Some studies
have reported that superoxide can lead to the translocation of Nrf2
from the cytoplasm to the nucleus, and sequentially binds to the
antioxidant responsive element (ARE) following SAH (8,9). Thus,
the Nrf2-ARE pathway has been significantly proven to play a
neuroprotective role in EBI after SAH, possibly via the inhibition
of cerebral oxidative stress by inducing antioxidant and
detoxifying enzymes (8,9).
Aloperine (ALO), an alkaloid isolated from
Sophora alopecuroides L. (Leguminosae), exerts a variety of
pharmacological activities. Previous studies have reported that the
protective functions of ALO can be attributed to its
anti-inflammatory, anticancer, anti-microbial, anti-viral and
anti-allergic effects (10–12). In addition, ALO has been reported to
exert significant neuroprotective effects against injury caused by
oxygen-glucose deprivation and reperfusion (OGD/RP) in
vitro, which can be attributed to its antioxidant capacity
(12). Significantly, Xu et
al (10) had demonstrated that
ALO could attenuate neuropathic pain induced by chronic
constriction injury via anti-oxidation activity in vivo.
Administration of ALO (80 and 40 mg/kg, intraperitoneally)
obviously increased the SOD concentration and reduced the level of
malondialdehyde (MDA) in the spinal cord. However, such antioxidant
mechanisms were not investigated in detail in earlier studies.
Considering the importance of antioxidant capacity for ALO, the
present study aimed to further evaluate the neuroprotective effect
of ALO in EBI after SAH and its relationship with the Nrf2-ARE
survival pathway.
Materials and methods
Experimental animals
In total, 150 adult male Sprague-Dawley rats
(250–300 g) were used in our study. Animals were purchased from the
Animal Department of Guizhou Medical University (Guiyang, China)
prior to surgery. Rats were housed under environmentally controlled
conditions in a 12/12 h light/dark cycle with a suitable
temperature of approximately 25°C, and provided with free access to
food and water. All experimental protocols were approved by the
Animal Care and Use Committee of Guizhou Medical University and
conformed to the Guide for the Care and Use of Laboratory Animals
by the National Institutes of Health.
SAH rat model
Briefly, the rats were anesthetized by an
intraperitoneal injection of 10% chloralic hydrate. The
experimental SAH model was induced by the stereotaxic insertion of
a needle into the pre-chiasmatic cistern in accordance with a
previous study (13). The needle was
placed 7.5 mm anterior to the bregma in the midline at an angle of
45° in the sagittal plane. Then, 0.3 ml of non-heparinized fresh
autologous arterial blood was slowly injected into the
pre-chiasmatic cistern for 20 sec with a pump using aseptic
technique. After injection, the burr hole was plugged immediately
by bone wax, and the rats were maintained in a 30° head-down
position for 30 min. Then, the rats were returned to their cages.
Sham animals were subjected to the same procedure exactly as
described above with the exception that no blood was injected
intra-cisternally.
Experimental design and drug
administration
All rats were randomly arranged into five groups as
follows: Sham group (n=30), SAH group (n=30), SAH+ vehicle group
(n=30), SAH+ low concentration ALO group (n=30), and SAH+ high
concentration group (n=30). ALO (Kmaels, Shanghai, China), was
dissolved in saline containing 10% acetic acid (2.5 mg/ml)
(14). It was then
intra-peritoneally administered to rats at 2 and 24 h after
induction of the SAH model (75 mg/kg in the low concentration group
and 150 mg/kg in the high concentration group) (10). Rats in the SAH+ vehicle group were
treated with 10% acetic acid in normal saline as above following
SAH insult. Analyses involving western blotting, TUNEL staining,
brain edema, immunohistochemistry (IHC), enzyme activity assays and
neurological deficits, used five rats from each experimental
group.
All animals were killed at a time-point 48 h after
SAH. The rats for Western blot analysis were infused
trans-cardially with 250 ml of cold heparinized 0.9% saline. Then,
the inferior basal temporal lobe tissue adjacent to the clotted
blood was dissected on ice, and immediately stored in liquid
nitrogen immediately until use. The rats for TUNEL staining and IHC
were perfused with 100 ml of ice-cold 0.9% saline through the left
ventricle and then fixed with 100 ml 4% paraformaldehyde. Then,
whole brains were removed and immersed in 4% paraformaldehyde and
stored at 4°C until use.
Western blot analysis
Briefly, tissues were mechanically lysed in 20 mM
Tris, pH 7.6 containing 0.2% sodium dodecyl sulfate (SDS), 1%
Triton X-100, 1% deoxycholate, 1 mM NaF, 2 mM
Na3VO4, and 1 mM phenylmethylsulphonyl
fluoride, and centrifuged at 12,000 × g for 15 min at 4°C for HO-1
and caspase-3 analysis. Nuclear protein was extracted for Nrf2
analysis as described previously (15). The concentration of each protein
sample was measured by DC protein assay (Beyotime, Nantong, China).
Equal amounts of protein per lane was separated by SDS-PAGE (10%
for Nrf2 and HO-1; 12% for caspase-3), and then transferred to a
polyvinylidene-difluoride (PVDF) membrane (Millipore, Darmstadt,
Germany). Membranes were then blocked with 5% skimmed milk for 2 h
at room temperature, and then incubated overnight at 4°C with
primary antibodies directed against Nrf2 (1:1,000 dilution; Abcam,
Cambridge, MA, USA), caspase-3 (1:1,000 dilution; Cell Signaling
Technology, Danvers, MA, USA), HO-1 (1:2,000 dilution; Abcam), and
GAPDH (1:5,000 dilution; Bioworld Technology, Inc., St. Louis Park,
MN, USA). After washing with Tween-TBS (4×10 min), the membranes
were incubated with goat anti-rabbit IgG-horseradish (1:5,000
dilution; Bioworld Technology, Inc.) secondary antibody for 2 h at
room temperature, then washed again (4×15 min) with Tween-TBS.
Finally, bands were visualized by enhanced ECL Western Blot
detection reagents (Millipore, Darmstadt, Germany), and exposed to
X-ray film (Kodak, Rochester, NY, USA). All experiments were
repeated at least three times.
IHC staining
Briefly, formalin-fixed paraffin-embedded sections
(4 µm) were prepared for IHC staining. Endogenous peroxidase
activity was blocked with 3% H2O2 for 10 min
followed by blocking serum for 1 h at room temperature. Sections
were then incubated with primary antibodies directed against Nrf2
(1:100 dilution) and HO-1 (1:200 dilution) (both from Abcam)
overnight at 4°C and then washed with PBS for 15 min. Afterwards,
the sections were incubated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (1:500; SantaS Cruz
Biotechnology, Santa Cruz, CA, USA) for 60 min at room temperature.
Diaminobenzidine (DAB) was then used as a chromogen and sections
were finally counterstained with haematoxylin. Sections incubated
in the absence of primary antibody were used as negative
controls.
Two slides (at least 250 µm apart) per rat were all
randomly selected from the cortex of the temporal lobe from each
rat brain. Positive cells were identified with the help of an
investigator who was blinded to the experimental groupings.
MDA level and GST-α1enzyme activity
assay
Frozen samples were homogenized in 10 mM Tris-HCl
(pH 7.8) and centrifuged for 15 min (12,000 × g) and the
supernatant collected for assay. MDA, a compound produced during
lipid peroxidation, was determined using the thiobarbituric acid
method. The given maximum absorbance was 535 nm. GST-α1 enzyme
activity was measured via the conjugation reaction of
1-chloro-2,4-dinitrobenzene and glutathione, calculating the slope
elevation of extinction coefficient at 340 nm (9).
TUNEL staining
TUNEL staining was performed following the
manufacturer's instructions (Roche Diagnostics, Basel,
Switzerland). Three slides (at least 200 µm apart) per rat were all
randomly selected. Positive cells were identified, counted, and
analyzed with the help of an experienced pathologist blinded to the
experimental groupings. The extent of brain impairment was
evaluated by apoptotic index, which was analyzed by the average
number of TUNEL positive cells in each section counted in 10
microscopic fields (at ×100 magnification).
Brain water content
Brains were collected at a time-point 48 h following
SAH. The left and right hemisphere brain tissues were immediately
weighed (wet weight) and then dried at a temperature of 100°C for
48 h to determine the dry weight. The percentage of water content
was calculated with the following formula: [(wet weight - dry
weight)/wet weight] ×100% (16).
Neurological deficit
Neurological deficit was performed 1 h before the
rats were euthanized at the 48 h time-point. Briefly, the
neurologic conditions of each rat, such as appetite, activity, and
walking deficit, were evaluated according to a scoring scale and
the average of the three grades was the neurological deficit. Each
rat's appetite was scored as 0, finished meal; 1, left meal
unfinished; 2, scarcely ate. Activity assessment was scored as 0,
walk and reach at least three corners of the cage; 1, walk with
some stimulation; 2, almost always lying down. Walking deficit was
scored as 0, no deficits; 1, unstable walk; 2, unable to walk
(17).
Statistical analysis
Data are expressed as mean ± SEM. All data were
analyzed by SPSS 17.0 software (SPSS Inc., Chicago, IL, USA), and
statistical differences among groups were compared by one-way
ANOVA, and differences between groups analysed by Tukey analysis.
Statistical significance was accepted when P<0.05.
Results
Mortality rates of the SAH rat model
48 h after induction
All the rats in our study survived the procedures of
experimental SAH or sham injury. There was no difference among
groups in terms of physiological parameters, including body
temperature, mean arterial blood pressure, or blood gas analysis
(data not shown). Mortality rate was 0 (0/30 rats) in the sham
group, 16.7% (6/36 rats) in the SAH group, 16.7% (6/36 rats) in the
SAH+ vehicle group, 14.3% (5/35 rats) in the low concentration ALO
group, and 11.8% (4/34 rats) in the high concentration ALO
group.
Western blot analysis for Nrf2, HO-1
and caspase-3 protein levels
Western blot analysis showed that the protein levels
of Nrf2 and HO-1 in the sham group were low and that levels were
both significantly increased in the SAH and SAH + vehicle groups in
comparison with the sham group (P<0.05, Fig. 1A). After ALO administration,
especially in the high concentration group, the protein levels of
Nrf2 and HO-1 were markedly elevated in comparison with the SAH+
vehicle group (P<0.05, Fig. 1A),
but not in the low concentration group (P>0.05, Fig. 1A).
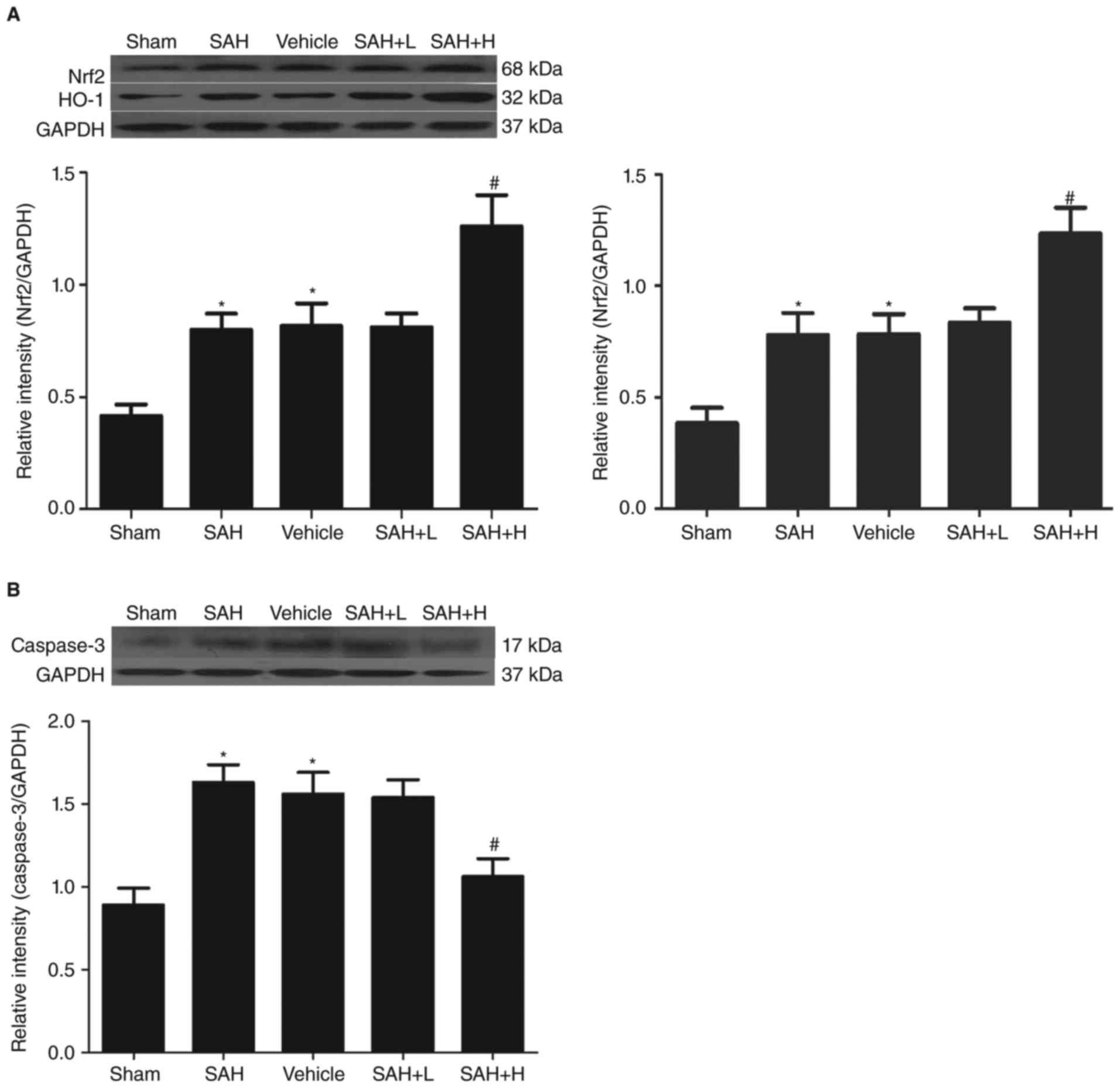 | Figure 1.Western blot analysis of Nrf2, HO-1
and caspase-3 levels in the cortex. (A) Different concentrations of
ALO influenced the levels of Nrf2, HO-1 at 48 h after SAH; (B)
different concentrations of ALO influenced the levels of caspase-3
at 48 h after SAH. SAH+ H, SAH+ high concentration ALO; SAH+ L,
SAH+ low concentration ALO; vehicle, SAH+ vehicle. *P<0.05 vs.
sham group, #P<0.05 vs. SAH+ vehicle group (n=5 per
group). Nrf2, nuclear factor erythroid-related factor 2; HO-1, heme
oxygenase-1; ALO, aloperine; SAH, subarachnoid hemorrhage. |
Similarly, compared with the sham group, the
expression of caspase-3 was markedly increased in the SAH and SAH +
vehicle groups, but this level was significantly downregulated
after high dose ALO treatment (P<0.05, Fig. 1B).
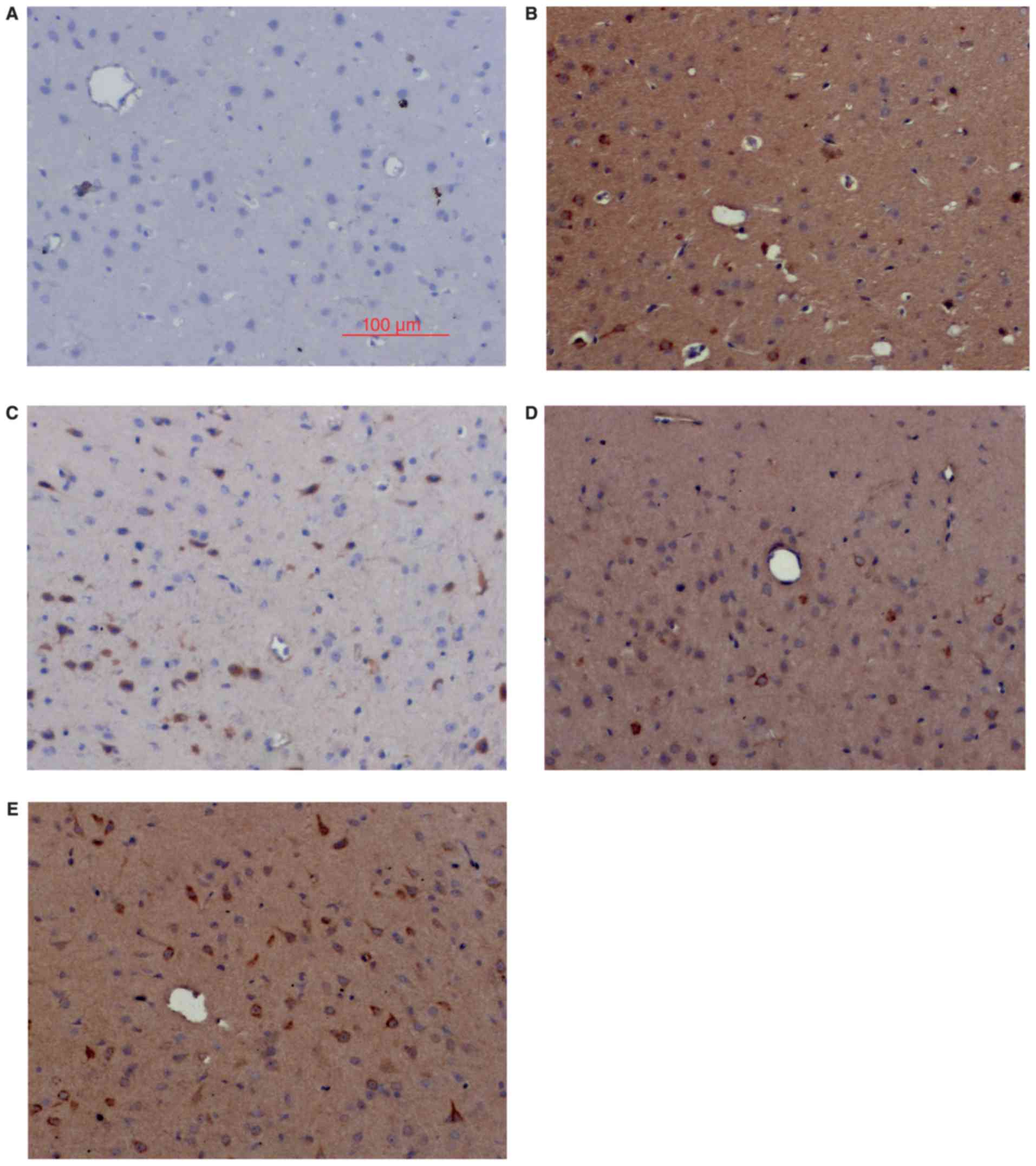
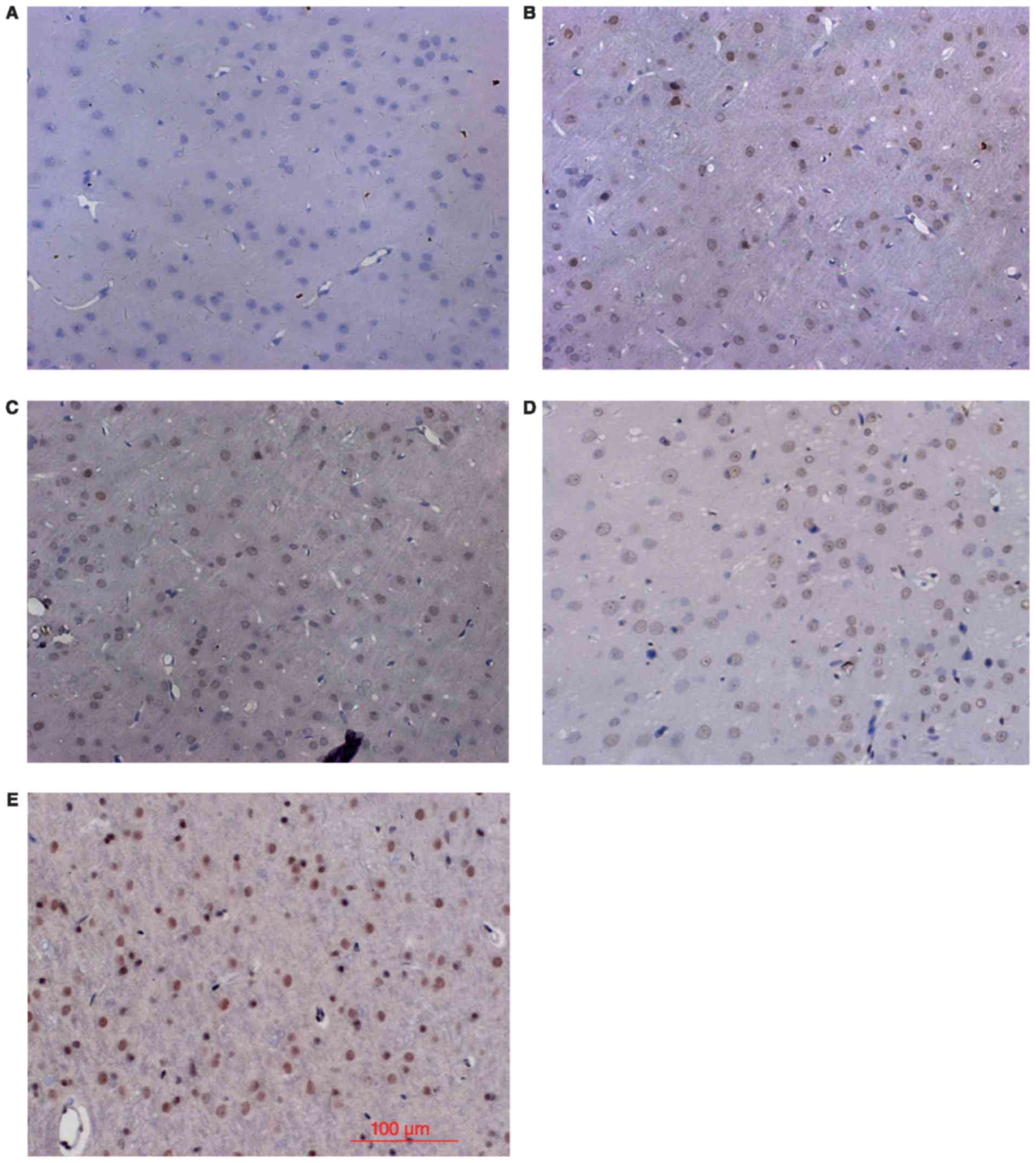
Nrf2 and HO-1 expression by IHC
Consistent with the western blot results, IHC
analysis showed little Nrf2 and HO-1 positive staining in the sham
group (Figs. 2 and 3), low levels of positive staining in the
SAH, SAH+ vehicle, and SAH+ low concentration ALO groups (Figs. 2B-D and 3B-D), but strong positive staining in the
SAH+ high concentration ALO group (Figs.
2E and 3E).
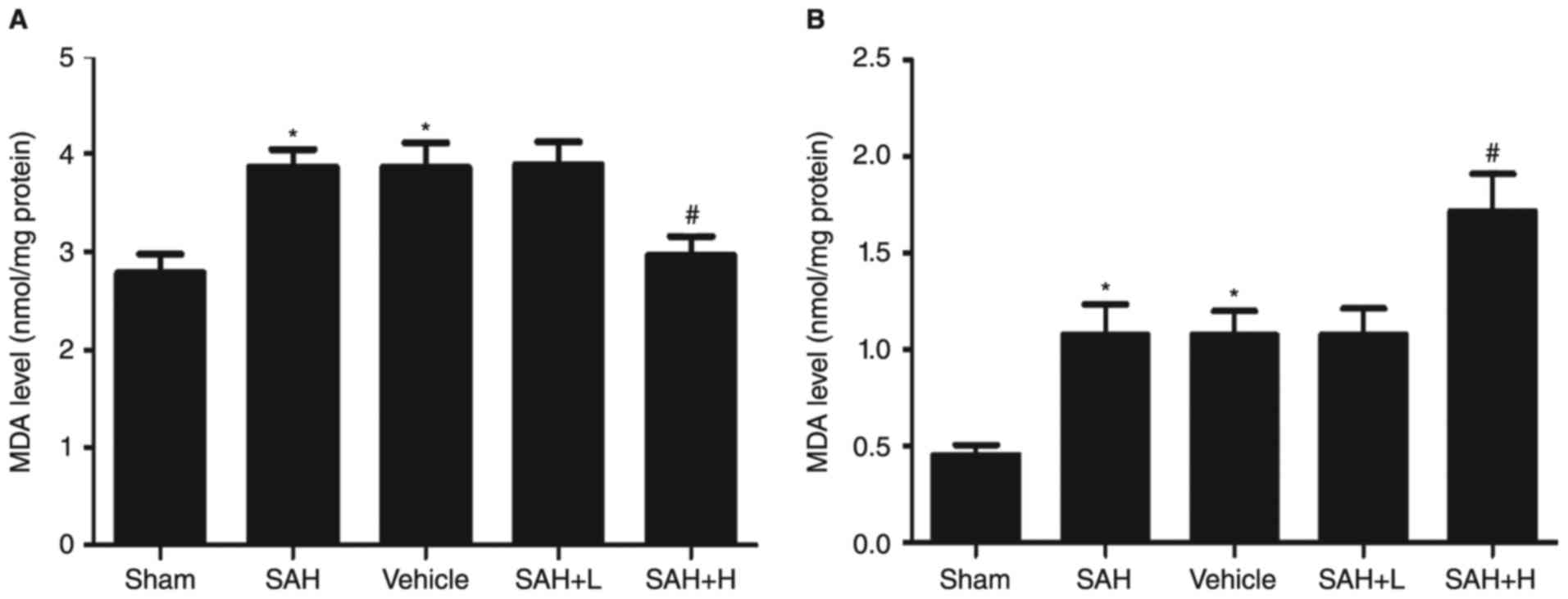
MDA levels and GST-α1enzyme activity
assay
Compared with the sham group, the levels of MDA in
the SAH, SAH + vehicle, and SAH+ low concentration ALO groups were
obviously increased (P<0.05, Fig.
4A). However, levels were significantly reduced with high
concentration ALO treatment in comparison with the SAH + vehicle
group (P<0.05, Fig. 4A).
Similarly, the enzyme activity of GST-α1 was upregulated following
SAH (P<0.05, Fig. 4B). After ALO
administration, especially in the high concentration group, the
activity was markedly elevated (P<0.05, Fig. 4B), but not in the low concentration
group (P>0.05, Fig. 4B).
Apoptotic death assays
TUNEL-positive apoptotic cells were observed in the
temporal lobe with deep brown staining in the nucleus. Few TUNEL
positive cells were found in the sham group, and apoptotic index
was low (Fig. 5). At 48 h after
initial bleeding, the apoptotic index was obviously increased in
the SAH and SAH+ vehicle groups in comparison with the sham group
(P<0.01, Fig. 5B, C and F). High
concentration ALO, but not low concentration ALO treatment,
significantly reduced apoptosis in SAH rats, as demonstrated by the
low apoptotic index in the SAH + high concentration ALO group
(P<0.05, Fig. 5D-F).
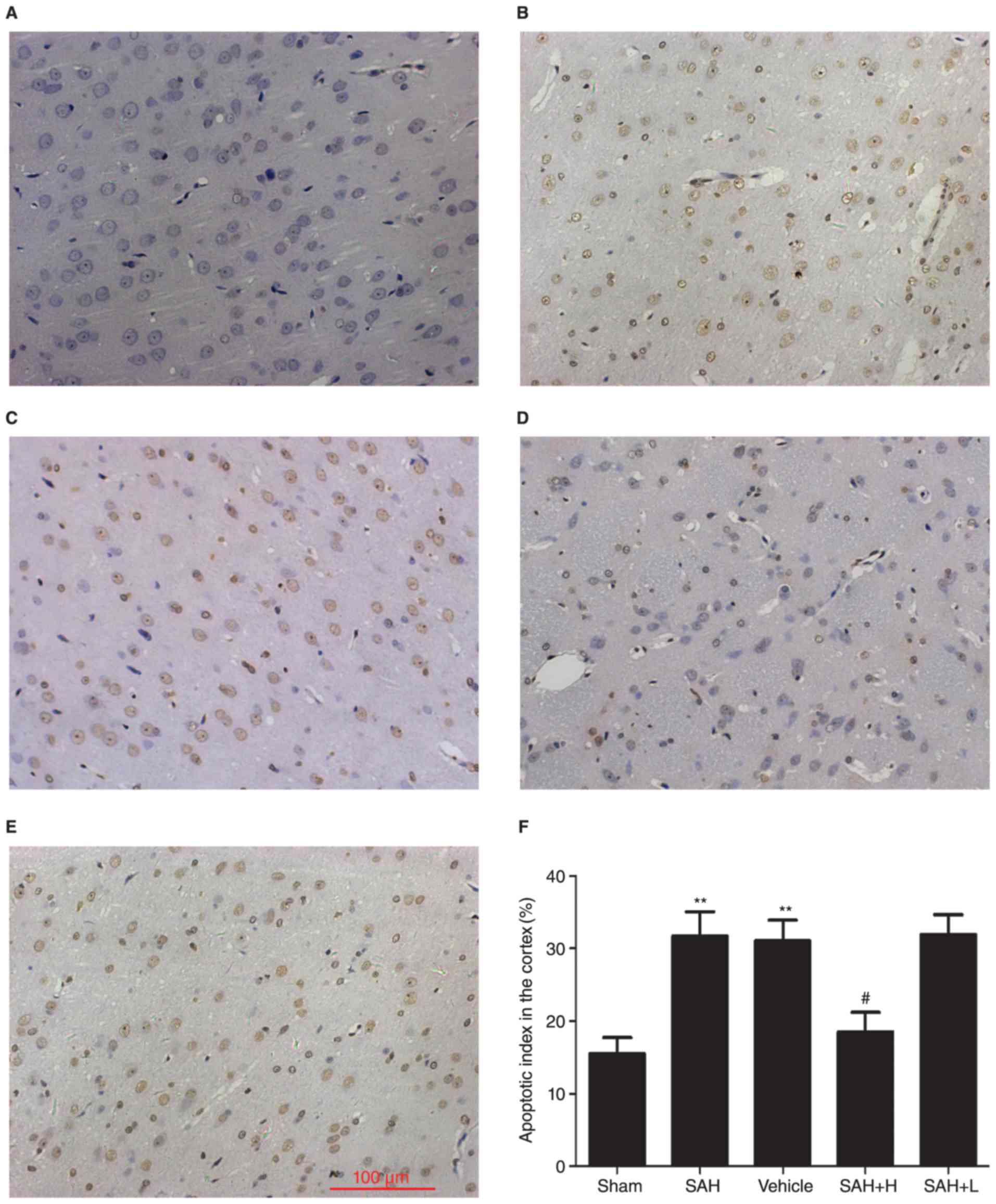 | Figure 5.Evaluation of neuronal apoptosis after
ALO treatment. (A) Sham group, (B) SAH group, (C) SAH+ vehicle
group, (D) SAH+ high concentration ALO group, (E) SAH+ low
concentration ALO group. (F) Quantification of apoptosis. Vehicle:
SAH+ vehicle; SAH+ H, SAH+ high concentration ALO; SAH+ L, SAH+ low
concentration ALO. **P<0.01 vs. sham group,
#P<0.05 vs. SAH+ vehicle group (scale bar, 100 µm,
n=5 per group). ALO, aloperine; SAH, subarachnoid hemorrhage. |
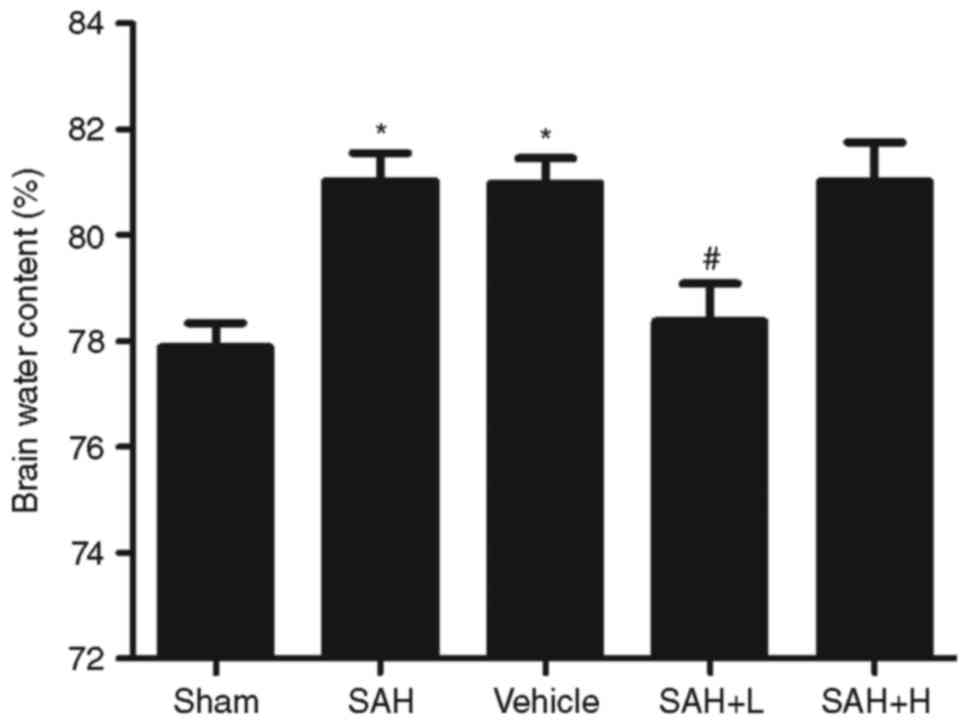
Brain edema
Compared with the sham group, obvious increments in
brain water content were detected at 48 h following SAH
(P<0.05, Fig. 6). The mean
value of brain water content was reduced after high concentration
ALO administration as compared with the SAH+ vehicle group
(P<0.05, Fig. 6).
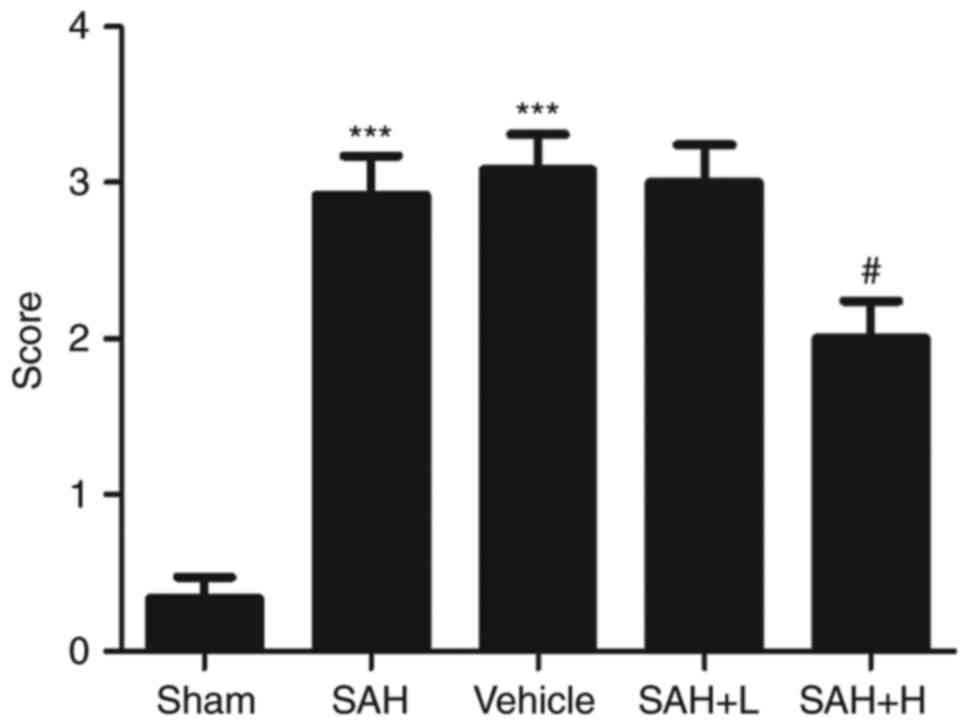
Neurological deficit
In comparison with the sham group, we found
significant impairment of neurological deficit in the SAH and SAH+
vehicle groups at 48 h after SAH (P<0.05; Fig. 7), and there was no difference between
the SAH and the SAH+ vehicle groups (P>0.05; Fig. 7). After the high concentration ALO
treatment, we observed a better level of neurological function. The
neurologic score of the high concentration ALO treatment group was
significantly lower than that of the SAH + vehicle group
(P<0.05; Fig. 7).
Discussion
Although it is well established that ALO can confer
a neuroprotective effect via attenuating oxidative stress in
vitro, our results further suggest that the antioxidative
effect of ALO might be attributed to activation of the Nrf2-ARE
survival pathway in EBI after SAH.
Factors responsible for the impact of the initial
bleeding in SAH include enhanced intracranial pressure (ICP),
reduced cerebral blood flow (CBF) and cerebral perfusion pressure
(CPP), BBB disruption, brain edema, acute vasospasm and dysfunction
of auto-regulation (18). Changes in
these factors constitute the EBI period. Despite extensive
research, the patient outcome post-aSAH remains poor. The fact that
the prevention of delayed cerebral vasospasm (dCVS) does not
improve clinical outcome suggests that its importance in patient
outcome has been misinterpreted (2).
Significantly, EBI has emerged as a new frontier and requires a
better understanding and consideration in elaborating therapeutic
strategy for improving aSAH outcome (1,2). In our
present paper, we created a modified rat model of SAH which has
been previously demonstrated to be suitable for pathological and
pathophysiological studies of SAH (19). The mortality rate of our SAH model
was acceptable, and that the experimental procedure is reproducible
and easy to perform. We were able to observe many TUNEL-positive
cells (Fig. 5B), an obvious
increment in brain edema (Fig. 6),
and upregulated caspase-3 levels (Fig.
1B) at 48 h after initial bleeding, which may partly be
ascribed to blood clotting, acute vasospasm, oxidative stress, or
acute ischemia injury occurring during the EBI period.
Although the exact mechanism of EBI remains obscure,
accumulating evidence has demonstrated that oxidative stress plays
an important role in the pathogenesis of EBI. Radicals can cause
damage to cardinal cellular components such as lipids, proteins,
and nucleic acids, which leads to subsequent cell death by modes of
necrosis or apoptosis, resulting in neuronal damage, cellular
apoptosis, endothelial injury, and BBB disruption (6,20). Such
damage can become even more widespread because of the weakened
cellular antioxidant defense systems. Furthermore, the
administration of systemic antioxidants in the experimental SAH
model has also been proven to reduce oxidative stress, protect the
BBB, alleviate apoptosis, and improve neurological scores (6,21).
Therefore, treatment with antioxidants may theoretically act to
prevent the propagation of tissue damage and improve both the
survival and neurological outcome of SAH. In addition, some
important studies have shown that the Nrf2-ARE pathway plays an
important role in antioxidant protection in various central nervous
system (CNS) diseases, such as cerebral ischemia, traumatic brain
injury (TBI), and SAH (9,15,19,22).
After initial bleeding, Nrf2 translocates into the nucleus and then
binds to ARE, activating a group of cytoprotective enzymes to
protect cells against oxidative damage. In the present study, we
similarly detected a significant increment of SAH-induced cerebral
expression and enzymatic activity of the ARE-mediated downstream
factors, HO-1, and GST-α1 (Figs. 1A
and 4B). These data were in
agreement with findings from a previous study illustrating the
activation of Nrf2-ARE pathway at 48 h following SAH (19).
ALO is a traditional Chinese medicine used for the
treatment of cancer. It has been reported to have versatile
biological effects, including anti-inflammatory, anti-apoptosis,
and anti-oxidation properties (10,12,23). In
the current study, we similarly detected that ALO treatment,
especially in the high dose group, significantly increased brain
Nrf2 and HO-1 levels, and inhibited oxidative stress in SAH rats in
comparison with rats in the SAH group (Figs. 1A and 4). Furthermore, statistical analysis
between the SAH and the SAH group given a high-concentration of ALO
revealed that ALO significantly ameliorated acute injury in EBI,
such as brain edema, apoptosis and neurological deficit (Figs. 5–7).
These results suggest that the acute administration of ALO could
confer an anti-oxidative effect in EBI after SAH, which might be
attributed to activation of the Nrf2-ARE survival pathway. Although
the anti-oxidative effects of ALO in acute injury have been
reported previously (12,14,23), the
molecular interaction between these factors is still unknown. Given
these diverse actions, it is likely that multiple signaling
pathways are involved in the protective effect of ALO in acute
injury. One vital study suggested that ALO was able to reduce the
content of MDA in the hepatic tissue of intoxicated animals but
without affecting Superoxide Dismutase (SOD) activity. In another
paper, Hu et al (14) found
that ALO could regulate activator protein-1 (AP-1) activity against
ischemia reperfusion (IR) -induced renal injury, and thus enhanced
SOD expression in order to increase ROS detoxification. These
discrepancies might be due to differences in the animal models
employed. Interestingly, in CNS studies, it has also been reported
that the protective mechanisms of ALO on cultured rat hippocampal
neurons injured by OGD/RP were related to anti-oxidative stress
(12). In this paper, Ma et
al (12) found that neurons in
the hippocampus from the ALO-group showed significantly reduced MDA
and increased endogenous anti-oxidant levels. In the present paper,
we detected a similar reduction in MDA level and increment in
GST-α1enzyme activity following ALO administration in EBI.
However, the mechanism underlying activation of the
Nrf2-ARE pathway by ALO has not been completely elucidated. Some
earlier studies have reported that several kinases can activate the
Nrf2-ARE pathway in response to some stimuli, including
phosphoinositol-3 kinase (PI3K) and extra cellular signal-regulated
protein kinase (ERK) (24,25). On the other hand, Hu et al
suggested that ALO protects mice against IR-induced renal injury by
regulating the PI3K pathway (14).
Herein, we speculate that ALO may play a role in the mechanisms
underlying activation of the Nrf2-ARE pathway against EBI after SAH
insult, which needs further investigation.
Although many compounds with neuroprotective effects
against acute injury are in various stages of pharmaceutical
development, ALO exhibits unique advantages. Sophora
alopecuroides L. and its related species have already
experienced thousands of years of human exposure with little
reported toxicity and side effects. In addition, ALO can easily
diffuse across biological membranes and the BBB in an energy
deficient environment (12).
Therefore, ALO may be a good candidate drug for the prevention and
treatment of SAH-induced acute injury.
It should be noted that there are some limitations
to our study. First, we did not explore the effect of ALO upon the
Nrf2-ARE pathway in the sham group. Second, the routes of ALO
administration usually include intraperitoneal injection,
intravenous injection, and oral administration. Here, we only
evaluated the effect of intraperitoneal injection. Third, although
the improved outcome was regarded as occurring partly by the
beneficial effects of ALO by activating the Nrf2-ARE pathway, Nrf2
knockout mice were not used in our study. In conclusion, our
results indicate that the acute administration of ALO can
ameliorate oxidative stress impairment in EBI, and that this
beneficial effect might involve activation of the Nrf2-ARE survival
pathway. Our study suggests that ALO may represent a new promising
therapeutic agent in the treatment of EBI after SAH.
References
|
1
|
Sehba FA, Hou J, Pluta RM and Zhang JH:
The importance of early brain injury after subarachnoid hemorrhage.
Prog Neurobiol. 97:14–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hou J and Zhang JH: Does prevention of
vasospasm in subarachnoid hemorrhage improve clinical outcome? No.
Stroke. 44 6 Suppl 1:S34–S36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cahill J, Calvert JW and Zhang JH:
Mechanisms of early brain injury after subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 26:1341–1353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsieh YP, Lin CL, Shiue AL, Yin H, Morrow
JD, Hsu JC, Hsieh TC, Wei HJ and Yen HC: Correlation of
F4-neuroprostanes levels in cerebrospinal fluid with outcome of
aneurysmal subarachnoid hemorrhage in humans. Free Radic Biol Med.
47:814–824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pyne-Geithman GJ, Caudell DN, Prakash P
and Clark JF: Glutathione peroxidase and subarachnoid hemorrhage:
Implications for the role of oxidative stress in cerebral
vasospasm. Neurol Res. 31:195–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gilgun-Sherki Y, Rosenbaum Z, Melamed E
and Offen D: Antioxidant therapy in acute central nervous system
injury: Current state. Pharmacol Rev. 54:271–284. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Itoh K, Tong KI and Yamamoto M: Molecular
mechanism activating Nrf2-Keap1 pathway in regulation of adaptive
response to electrophiles. Free Radic Biol Med. 36:1208–1213. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Zhu Y, Zhou D, Wang Z and Chen G:
Recombinant human erythropoietin (rhEPO) alleviates early brain
injury following subarachnoid hemorrhage in rats: Possible
involvement of Nrf2-ARE pathway. Cytokine. 52:252–257. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu Q, Zhang XS, Wang HD, Zhang X, Yu Q, Li
W, Zhou ML and Wang XL: Astaxanthin activates nuclear factor
erythroid-related factor 2 and the antioxidant responsive element
(Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in
rats and attenuates early brain injury. Mar Drugs. 12:6125–6141.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu YQ, Jin SJ, Liu N, Li YX, Zheng J, Ma
L, Du J, Zhou R, Zhao CJ, Niu Y, et al: Aloperine attenuated
neuropathic pain induced by chronic constriction injury via
anti-oxidation activity and suppression of the nuclear factor kappa
B pathway. Biochem Biophys Res Commun. 451:568–573. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao P, Zhou R, Zhu XY, Hao YJ, Li N, Wang
J, Niu Y, Sun T, Li YX and Yu JQ: Matrine attenuates focal cerebral
ischemic injury by improving antioxidant activity and inhibiting
apoptosis in mice. Int J Mol Med. 36:633–644. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma NT, Zhou R, Chang RY, Hao YJ, Ma L, Jin
SJ, Du J, Zheng J, Zhao CJ, Niu Y, et al: Protective effects of
aloperine on neonatal rat primary cultured hippocampal neurons
injured by oxygen-glucose deprivation and reperfusion. J Nat Med.
69:575–583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ansar S and Edvinsson L: Subtype
activation and interaction of protein kinase C and
mitogen-activated protein kinase controlling receptor expression in
cerebral arteries and microvessels after subarachnoid hemorrhage.
Stroke. 39:185–190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu S, Zhang Y, Zhang M, Guo Y, Yang P,
Zhang S, Simsekyilmaz S, Xu JF, Li J, Xiang X, et al: Aloperine
protects mice against ischemia reperfusion (IR)-induced renal
injury by regulating PI3K/AKT/mTOR signaling and AP-1 activity. Mol
Med. Nov 3–2015.(Epub ahead of print). View Article : Google Scholar
|
|
15
|
Yan W, Wang HD, Feng XM, Ding YS, Jin W
and Tang K: The expression of NF-E2-related factor 2 in the rat
brain after traumatic brain injury. J Trauma. 66:1431–1435. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng G, Chunlei W, Pei W, Zhen L and
Xiangzhen L: Simvastatin activates Akt/glycogen synthase
kinase-3beta signal and inhibits caspase-3 activation after
experimental subarachnoid hemorrhage. Vascul Pharmacol. 52:77–83.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamaguchi M, Zhou C, Nanda A and Zhang JH:
Ras protein contributes to cerebral vasospasm in a canine
double-hemorrhage model. Stroke. 35:1750–1755. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ostrowski RP, Colohan AR and Zhang JH:
Molecular mechanisms of early brain injury after subarachnoid
hemorrhage. Neurol Res. 28:399–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Ma C, Meng CJ, Zhu GQ, Sun XB, Huo
L, Zhang J, Liu HX, He WC, Shen XM, et al: Melatonin activates the
Nrf2-ARE pathway when it protects against early brain injury in a
subarachnoid hemorrhage model. J Pineal Res. 53:129–137. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ayer RE and Zhang JH: Oxidative stress in
subarachnoid haemorrhage: Significance in acute brain injury and
vasospasm. Acta Neurochir Suppl. 104:33–41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ayer RE and Zhang JH: The clinical
significance of acute brain injury in subarachnoid hemorrhage and
opportunity for intervention. Acta Neurochir Suppl. 105:179–184.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ren J, Fan C, Chen N, Huang J and Yang Q:
Resveratrol pretreatment attenuates cerebral ischemic injury by
upregulating expression of transcription factor Nrf2 and HO-1 in
rats. Neurochem Res. 36:2352–2362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang H, Yang S, Zhou H, Sun M, Du L, Wei
M, Luo M, Huang J, Deng H, Feng Y, et al: Aloperine executes
antitumor effects against multiple myeloma through dual apoptotic
mechanisms. J Hematol Oncol. 8:262015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zipper LM and Mulcahy RT: Erk activation
is required for Nrf2 nuclear localization during pyrrolidine
dithiocarbamate induction of glutamate cysteine ligase modulatory
gene expression in HepG2 cells. Toxicol Sci. 73:124–134. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ku BM, Joo Y, Mun J, Roh GS, Kang SS, Cho
GJ, Choi WS and Kim HJ: Heme oxygenase protects hippocampal neurons
from ethanol-induced neurotoxicity. Neurosci Lett. 405:168–171.
2006. View Article : Google Scholar : PubMed/NCBI
|