Introduction
Colon cancer, a frequently diagnosed cancer type
worldwide, is a disease in which malignant tumors form in the
tissues of the colon and it is one of the leading causes of
cancer-related mortality worldwide (1). Currently, the two main options for
treatment of colon cancer are chemotherapy and surgery, and the
treatment option is dependent upon the tumor location, size and
stage of cancer, and overall characteristics of the patient
affected (2,3). Chemotherapy may be given at any stage
and is generally administered following surgery; however, in some
cases it is also given prior to surgery in order to reduce the
tumor size (4). The overall survival
of patients with colon cancer has increased over the past decade
due to improvements in medical sciences and chemotherapeutic
regimens (5,6). Despite this, almost all patients with
colon cancer develop drug resistance, which decreases the efficacy
of the drugs and ultimately leads to failure of chemotherapy
(7,8).
Decreases in the effect of drugs, including
antibiotics or chemotherapeutic agents, such as doxorubicin, is
termed drug resistance (9,10). Treating drug resistance, particularly
multidrug resistance, is one of the major obstacles to successful
chemotherapy (11). Furthermore, the
majority of cancer-related mortalities occur due to failure of
chemotherapy that occurs due to the generation of drug resistance
during the process of chemotherapy and cancer progression (12). Evidence suggests that cancer cell
resistance to chemotherapy is due to a transition from an
epithelial to mesenchymal phenotype (13). Other research has demonstrated an
association of epithelial to mesenchymal transition (EMT) with
acquired resistance to cancer therapy, as well as cancer metastasis
(14). As an outcome of these
findings, if the epithelial phenotype is restored, it increases the
sensitivity of tumor cells to chemotherapy (15). EMT of tumor cells not only causes
increased metastasis, but also contributes to drug resistance
(16). Intricate links between cells
with an EMT-like phenotype and drug resistance in tumors have also
been proposed (17). A diverse array
of cytokines and growth factors may contribute to the regulation of
the process of drug resistance development (16,18). The
development of drug resistance may also be regulated by a higher
apoptotic threshold, aerobic glycolysis, regions of hypoxia and
elevated activity of drug efflux transporters (19). Furthermore, a previous report has
indicated that drug resistance emergence may occur as a consequence
of EMT (16). Evidence for an
eminent role of steroid receptor coactivator (SRC) in invasion and
in other tumor progression-related events, such as EMT, also exists
(19). There are several receptors,
as well as non-receptor tyrosine kinases (RTKs), which are
modulated by SRC and are responsible for persistence and robustness
of RTK signaling (20). It is
important to study the mechanism underlying drug resistance in
cancer and the ways by which drug resistance may be reversed.
Over the past few years, multiple mechanisms
responsible for drug resistance have been proposed by different
groups, which may be broadly divided into two types: Cellular and
non-cellular resistance mechanisms (21,22).
Cellular mechanisms include enzymes, targets of drugs and inside
transport systems of cancer cells, whereas non-cellular mechanisms
include extracellular factors, such as the microenvironment of the
tumor and vascular accessibility (23–25). The
present study focused on the generation of a drug-resistant cell
line and the ways by which the chemoresistance may be reversed
(26). The present study was able to
successfully demonstrate that, instead of targeting multiple
signaling pathways that are activated in drug resistance, it is
possible to target a common signaling node that aids in the
reversal of drug resistance.
Materials and methods
Drugs, reagents and chemicals
Doxorubicin, saracatinib and triciribine were
purchased from Selleck Chemicals (Shanghai, China). RPMI-1640,
radioimmunoprecipitation assay (RIPA) buffer, Hanks buffer, MTT and
Bradford reagent were obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Probes, SuperScript™ One-Cycle cDNA
kit cDNA kits for reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) and the following primers were obtained
from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA):
E-cadherinP-F, 5′-GGGGTACCTGTCTCTCTACAAAAAGGCA-3′ and
E-cadherinP-R, 5′-GGAAGATCTGGGCTGGAGCGGGCTGGAGT-3′; epithelial cell
adhesion molecule (EpCAM)P-F, 5′-CGCAGCTCAGGAAGAATGTG-3′ and
EpCAMP-R, 5′-TGAAGTACACTGGCATTGACG-3′; vimentinP-F,
5′-GGCTCAGATTCAGGGGAACAGC-3′ and vimentinP-R,
5′-CAGGTTGTGCAGGTTGTTCTA-3′; N-cadherinP-F,
5′-CACTGCTCAGGACCCAGAT-3′ and N-cadherinP-R,
5′-TAAGCCGAGTGATGGTCC-3′; GAPDHP-F, 5′-GGTGTGAACGGATTTGGCCGTATTG-3′
and GAPDHP-R, 5′-CCGTTGAATTTGCCGTGAGTGGAGT-3′. Fetal bovine serum
(FBS), Opti-MEM (cat. no. 11058021) and Anti-Anti were procured
from Gibco (Thermo Fisher Scientific, Inc.). FuGENE 6 (cat. no.
PRE2691) was obtained from Promega Corporation (Madison, MI, USA).
Phosphorylated (p)-SRC 416 (10% SDS-PAGE; cat. no. 2101; dilution
1:1,000), AKT (10% SDS-PAGE; cat. no. 9272; dilution 1:1,000),
phosphatase and tensin homolog (PTEN; 10% SDS-PAGE; cat. no. 9188;
dilution 1:1,000), epidermal growth factor receptor (EGFR; 6%
SDS-PAGE; cat. no. 4267; dilution 1:1,000), p-SRC 416 (10%
SDS-PAGE; cat. no. 2101; dilution 1:1,000), insulin-like growth
factor 1 receptor (IGF-1R; 8% SDS-PAGE; cat. no. 9750; dilution
1:1,000), p-AKT ser473 (10% SDS-PAGE; cat. no. 4060; dilution
1:1,000), SRC (10% SDS-PAGE; cat. no. 2109; dilution 1:1,000),
E-cadherin (6% SDS-PAGE; cat. no. 14472; dilution 1:1,000), EpCAM
(12% SDS-PAGE; cat. no. 2929; dilution 1:1,000), N-cadherin (8%
SDS-PAGE; cat. no. 13116; dilution 1:1,000), vimentin (10%
SDS-PAGE; cat. no. 5741; dilution 1:1,000), β-actin (cat. no. 4970;
dilution 1:2,000), anti-rabbit secondary antibody (cat. no. 93702;
dilution 1:2,500), anti-mouse secondary antibody (cat. no. 14709;
dilution 1:2,500), small interfering (si)RNA control (cat. no.
6568; dilution 100 nM), siRNA EGFR (cat. no. 6480; dilution 200 nM)
and siRNA insulin-like growth factor receptor (IGFR; cat. no. 6610;
dilution 200 nM) were obtained from Cell Signaling Technology Inc.
(Danvers, MA, USA). shRNA control (cat. no. sc-108060; dilution 1
µg), shRNA EGFR (cat. no. sc-29301; dilution 1 µg) and shRNA IGFR
(cat. no. sc-35638; dilution 1 µg) were obtained from Santa Cruz
Biotechnology Inc. (Dallas, TX, USA).
Cell line and culture conditions
The LS180 cell line was purchased from American Type
Culture Collection (Manassas, VA, USA), cultured in RPMI-1640
supplemented with 10% FBS and 1% antibiotic Anti-Anti and grown in
an incubator at 37°C with 5% CO2 and 95% humidity. For
generation of doxorubicin resistance, LS180 cells were continuously
treated with the drug (from 10 nM up to 30 µM) for a period of 6
months in an incubator at 37°C containing 5% CO2 and 95%
humidity. Prior to two days of subsequent experiments, resistant
cells were grown in media without the addition of any drug. For
overexpression/knockdown, transient transfection was performed.
Cells were grown in six-well plates at a density of
1×106 cells in transfection media (Opti-MEM). The
transfection mixture contains 35 µl of FuGENE 6 and siRNA (1:300)
or shRNA (1:200) was prepared and incubated for 30 min prior to
addition to the cells. After 24 h of transfection, cells were
washed with PBS and lysates were prepared for western blot
analysis.
Cell viability assay
The parental and doxorubicin-resistant LS180 cell
lines were seeded at a density 1.5×104 cells/well and
allowed to grow for 24 h to attain morphology and stationary state.
After 24 h, both parental and doxorubicin-resistant cells were
treated with doxorubicin at a concentration of 2, 5, 10, 20, 40, 80
and 100 µM in an incubator containing 5% CO2 at at 37°C
and 95% humidity for 12 and 24 h. MTT was added at a concentration
of 2.5 mg/ml to each well before 4 h of termination. Finally, the
formazan crystals were dissolved in dimethyl sulfoxide and
absorbance was measured at 570 nm using a synergy MX plate reader
(BioTek Instruments, Inc., Winooski, VT, USA).
Western blotting
Parental and resistant LS180 cells were seeded in
60-mm dishes at a density of 1.5×106 cells for 24 h.
After 24 h, the cells were treated with different inhibitors (1 µM
doxorubicin, 1 µM saracatinib and 1 µM triciribine). These cells
were lysed in RIPA buffer and protein estimation was performed
using the Bradford method. Proteins (70 µg) were separated on 10%
SDS-PAGE and transferred onto a nitrocellulose membrane at 100 V
for 2 h. Membranes were blocked in 5% fat-free skimmed milk for 1 h
at room temperature to avoid non-specific binding. After 1 h,
primary antibodies were added and incubated overnight at 4°C.
Protein blots were washed with Tris-buffered saline-Tween-20 twice
for 5 min each. Subsequently, horseradish peroxidase-conjugated
secondary antibody was added at room temperature for 1 h, followed
by washing thrice with blocking buffer for 5 min each. Finally,
using an enhanced chemiluminescence kit (GE Healthcare, Chicago,
IL, USA), the bands of proteins were analyzed on an X-ray film.
Quantification of all western blots were was performed by
normalized by β-actin and using Image J software (v.1.48, National
Institutes of Health, Bethesda, MD, USA).
Colony formation assay
Doxorubicin-resistant LS180 cells were seeded in a
six-well plate and allowed to grow for 24 h. After 24 h, these
cells were treated with 1 µM doxorubicin and 1 µM saracatinib alone
and in combination in an incubator at 37°C with 5% CO2
and 95% humidity for 48 h. Following this, cells were trypsinized
and replated in a six-well plate at a density of 500 cells/well and
allowed to grow for 21 days. On experiment termination, cells were
first washed thrice with PBS and then fixed in 4% paraformaldehyde
for 10–15 min at room temperature. Crystal violet (0.06%) at room
temperature was used to stain live cells at 25°C for 10 min and
images were captured using a light microscope at a magnification of
×30 (Olympus Corporation, Tokyo, Japan).
Three-dimensional (3D) sphere
formation assay
Using MammoCult medium (Stemcell Technologies, Inc.,
Vancouver, BC, USA), single Doxo LS180 cells were seeded in
ultra-low attachment plates at a density of 1×105
cells/well and allowed to grow for 7 days. Following this, the
cells were treated with doxorubicin (1 µM) and saracatinib (1 µM)
alone and in combination for 48 h. Subsequent to treatment, the
primary spheres were dissociated by pipetting and single cells were
replated in ultra-low attachment 6-well plates at a density of
5×104 cells/well. Secondary spheres were counted using a
light microscope at a magnification of ×30 after 21 days of
incubation.
mRNA quantification
Parental and doxorubicin-resistant cells were
cultured and total RNA was isolated using TRIzol reagent (cat. no.
T9424 obtained from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
RNA was purified using an RNeasy mini kit (Qiagen China Co., Ltd.,
Shanghai, China). Purified RNA was first used to generate cDNA
using an M-MLV RT kit (Promega Corporation, Madison, WI, USA)
according to the instructions of manufacturer and then qPCR
analysis was conducted using a TaqMan universal PCR master mix
(Roche Applied Science, Penzberg, Germany). Reverse transcription
was performed according to the following thermocycling conditions:
Denaturation at 94°C for 30 sec and annealing and elongation at
72°C for 1 min, followed use of the aforementioned primers on an
ABI PRISM sequencing detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) The relative fold change of differential
inducible expression of the genes vs. control group was quantified
by using 2−∆∆Cq method (27).
Statistical analysis
GraphPad Instat3 software (GraphPad software Inc.,
La Jolla, CA, USA) was used for statistical analysis. All
experiments were performed three times and values of each
experiment were demonstrated as the mean ± standard deviation. For
comparison of each experiment, one-way analysis of variance was
performed followed by Bonferroni's test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Development of a chemoresistant cell
line
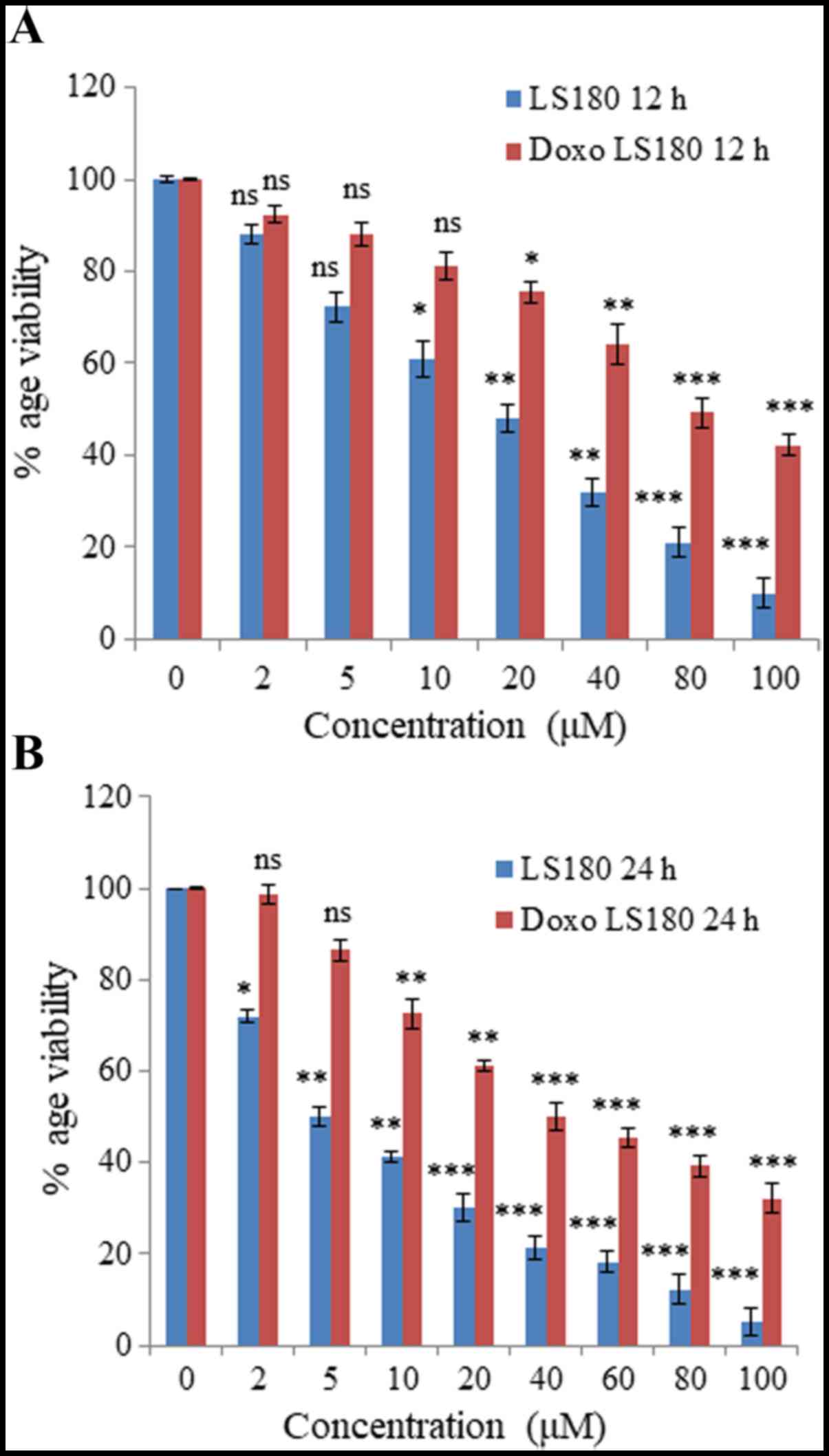
Colon cancer cell line LS180 was selected to
generate resistance towards doxorubicin. Following attainment of
80% confluency, cells were treated with doxorubicin beginning with
a low dose of 50 nM and treatment was retained continuously until
the growth of cells began to decrease and they demonstrated
morphological changes. The resistance of these cells was obtained
in 6 months and was confirmed by an MTT assay. The IC50
of doxorubicin-resistant cells was 80 µM whereas the parental
cancer cell line was 18 µM at 12 h (Fig.
1A). Notably, IC50 of doxorubicin in parental cell
line was 5 µM, whereas in resistant cells the IC50 was
increased up to 40 µM at 24 h (Fig.
1B).
Doxorubicin resistance induces an
EMT-like phenotype
For generation of a stable doxorubicin-resistant
cell line, LS180 cells were continuously treated with doxorubicin
for 6 months. After 6 months, there was a marked difference in the
morphology of parental and resistant cells. Resistant cells were
mesenchymal in shape (analyzed by microscopy; data not shown).
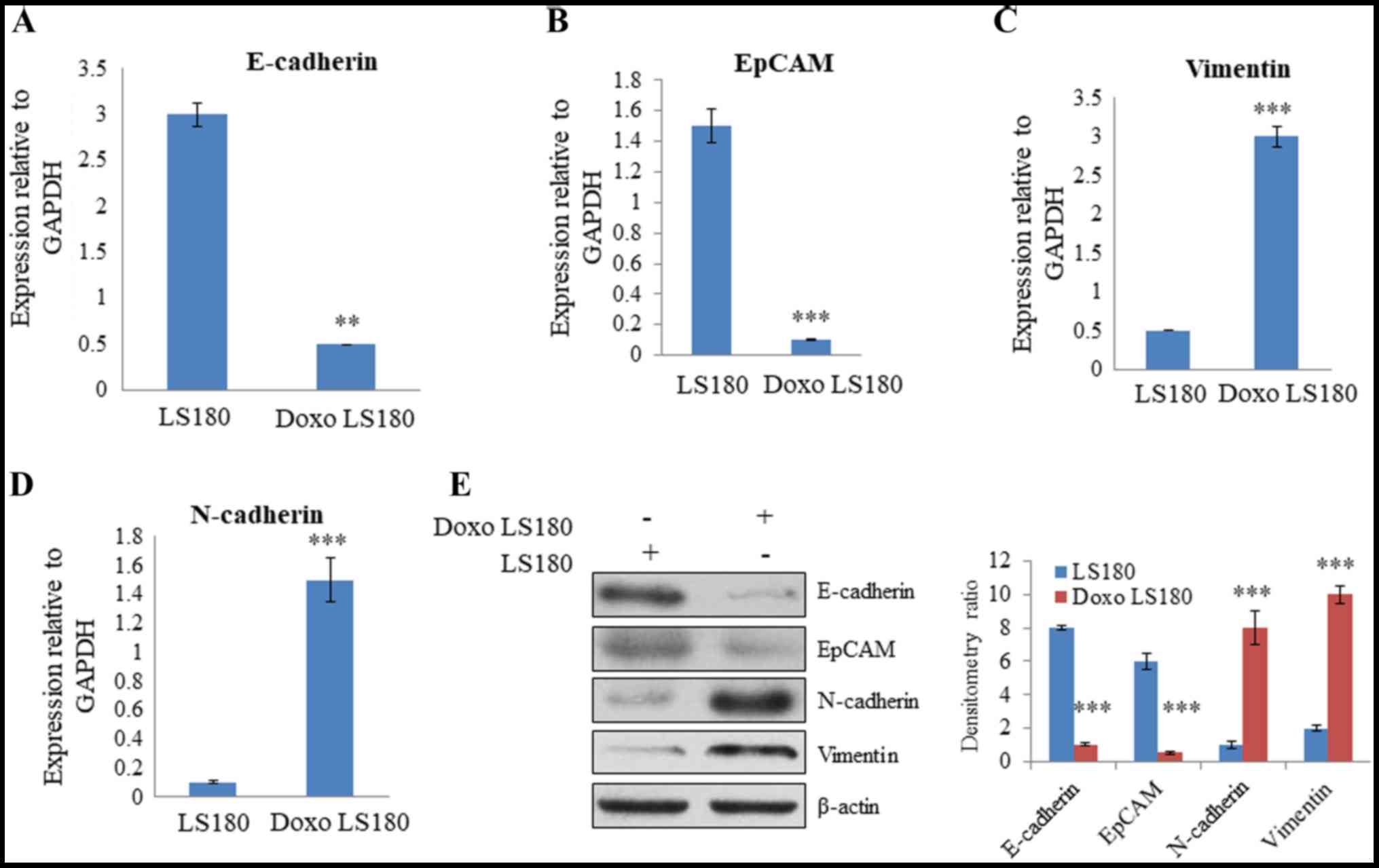
RT-qPCR analysis of EMT-related genes, including N-cadherin,
vimentin, EpCAM and E-cadherin, was performed to further analyze
the difference between parental and resistant cells. The results
revealed that there was a significant decrease in the expression of
E-cadherin and EpCAM, two epithelial markers, in resistant cells
compared with the level in parental cells (Fig. 2A and B). Furthermore, there was a
significant increase in the expression level of mesenchymal
markers, vimentin and N-cadherin, in the doxorubicin-resistant
cells compared with the levels observed in the parental cells
(Fig. 2C and D). Western blotting
demonstrated that E-cadherin and EpCAM were expressed at a higher
level in parental cells than in resistant cells. However, the
expression of mesenchymal markers, N-cadherin and vimentin, were
significantly increased in cells resistant to doxorubicin, and the
epithelial markers, E-cadherin and EpCAM, demonstrated reduced
expression levels compared with the levels in parental cells
(Fig. 2E). Together these results
indicate that doxorubicin leads to resistance in colon cancer and
induces an EMT-like phenotype.
SRC is activated in
doxorubicin-resistant cells
Cancer cells are known to be evolved that undergo
reprogramming and become drug-resistant (28). In resistant LS180 cells, SRC was
observed to be activated (indicated by phosphorylation at position
416) compared with the parental cell line (Fig. 3A). However, there was no notable
change in the protein expression levels of PTEN and AKT, which
further confirmed that resistance was due to doxorubicin and not
due to the presence of any pre-existing phosphoinositide 3-kinase
mutation (Fig. 3A).
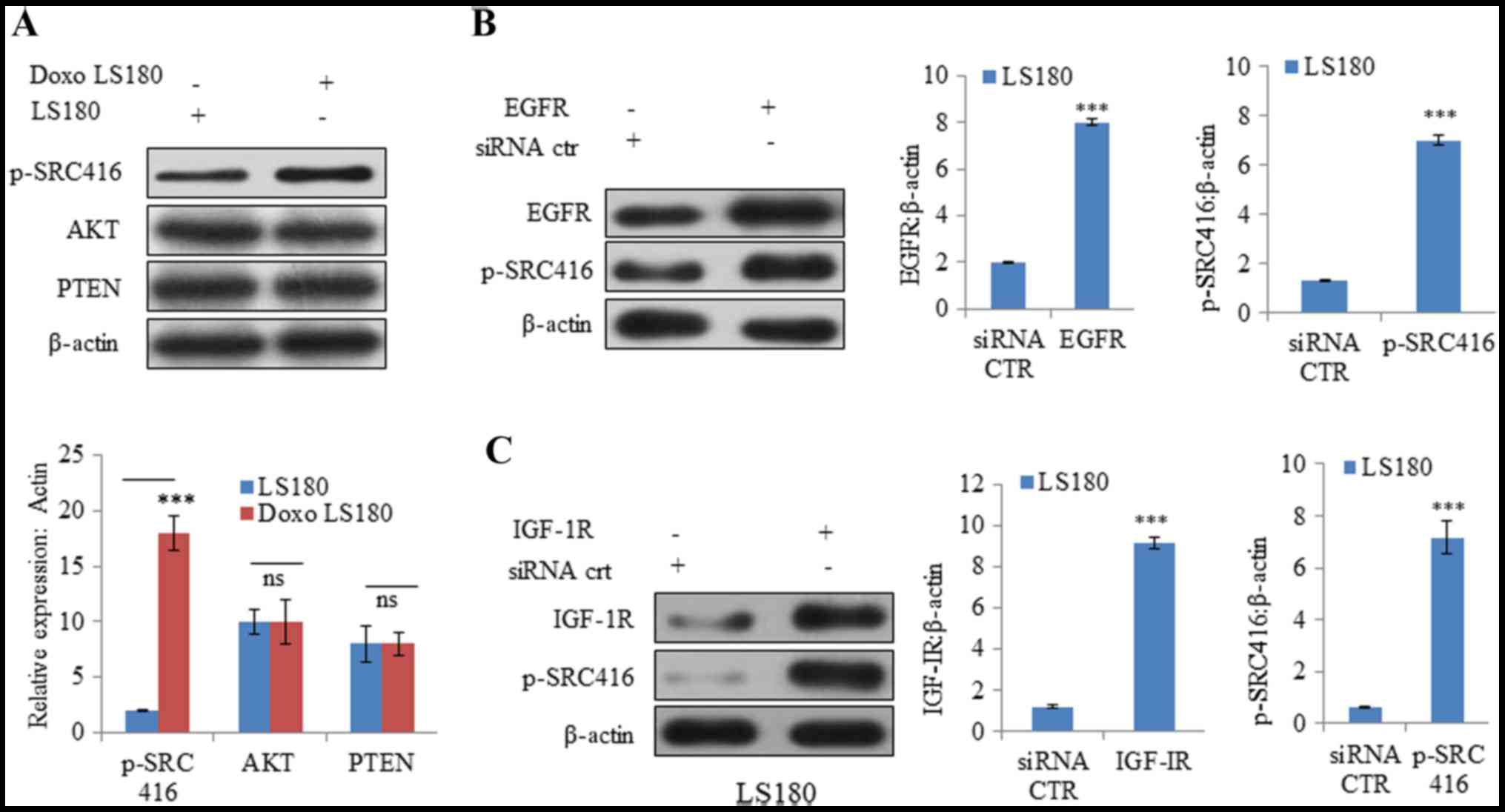 | Figure 3.SRC is activated by Doxo resistance.
(A) Doxo resistance induces activation of oncogenic protein SRC
independent of AKT and PTEN, as demonstrated by increased
phosphorylation of SRC at Y416 with no notable effect on AKT and
PTEN. Overexpression of receptor tyrosine kinases (B) EGFR
activates SRC activation in Doxo LS180 and (C) IGF-1R also resulted
SRC activation in LS180 cells. Knockdown of tyrosine kinases (D)
EGFR and (E) IGF-1R in Doxo-resistant cells led to decreased
phosphorylation of SRC. Data is presented as the mean ± standard
deviation of three different experiments. Statistical comparisons
were made using Bonferroni's method ***P<0.001 vs. siRNA CTR;
$$P<0.01 and $$$P<0.001 vs. shRNA CTR.
SRC, steroid receptor coactivator; Doxo, doxorubicin; Doxo LS180,
doxorubicin-resistant LS180 cells; PTEN, phosphatase and tensin
homolog; EGFR, epidermal growth factor receptor; IGF-1R,
insulin-like growth factor 1 receptor; p, phosphorylated; siRNA,
small interfering RNA; sh, short hairpin; CTR, control. |
Subsequently, the roles of other RTKs, including
EGFR and IGF-1R, were examined in SRC activation. Overexpression of
EGFR and IGF-1R resulted in increased phosphorylation at position
416, indicating SRC activation (Fig. 3B
and C). Furthermore, knockdown of EGFR and IGF-1R in
doxorubicin-resistant cells resulted in SRC inactivation (decreased
phosphorylation of SRC at position 416; Fig. 3D and E).
Activation of RTKs by SRC induces
doxorubicin resistance
To understand the mechanism of doxorubicin-induced
SRC activation in resistant cells, SRC activity was inhibited by
using a small molecule SRC inhibitor, saracatinib, as well as SRC
siRNA. It was observed that AKT phosphorylation was significantly
inhibited by doxorubicin in the parental cell line, whereas
doxorubicin-resistant cells were resistant towards
doxorubicin-mediated inhibition of AKT (Fig. 4A). However, when resistant cells were
treated with SRC inhibitor saracatinib and SRC shRNA in combination
with doxorubicin, there was a significant decrease in the
phosphorylation of AKT compared with non-treated cells (Fig. 4B). In EGFR and IGF-1R overexpressing
cells, SRC inhibition led to significant inhibition of AKT compared
with non-treated cells (Fig. 4C).
These results suggest that different RTKs, along with their
downstream targets that are activated due to drug resistance, may
be effectively blocked by inhibiting SRC.
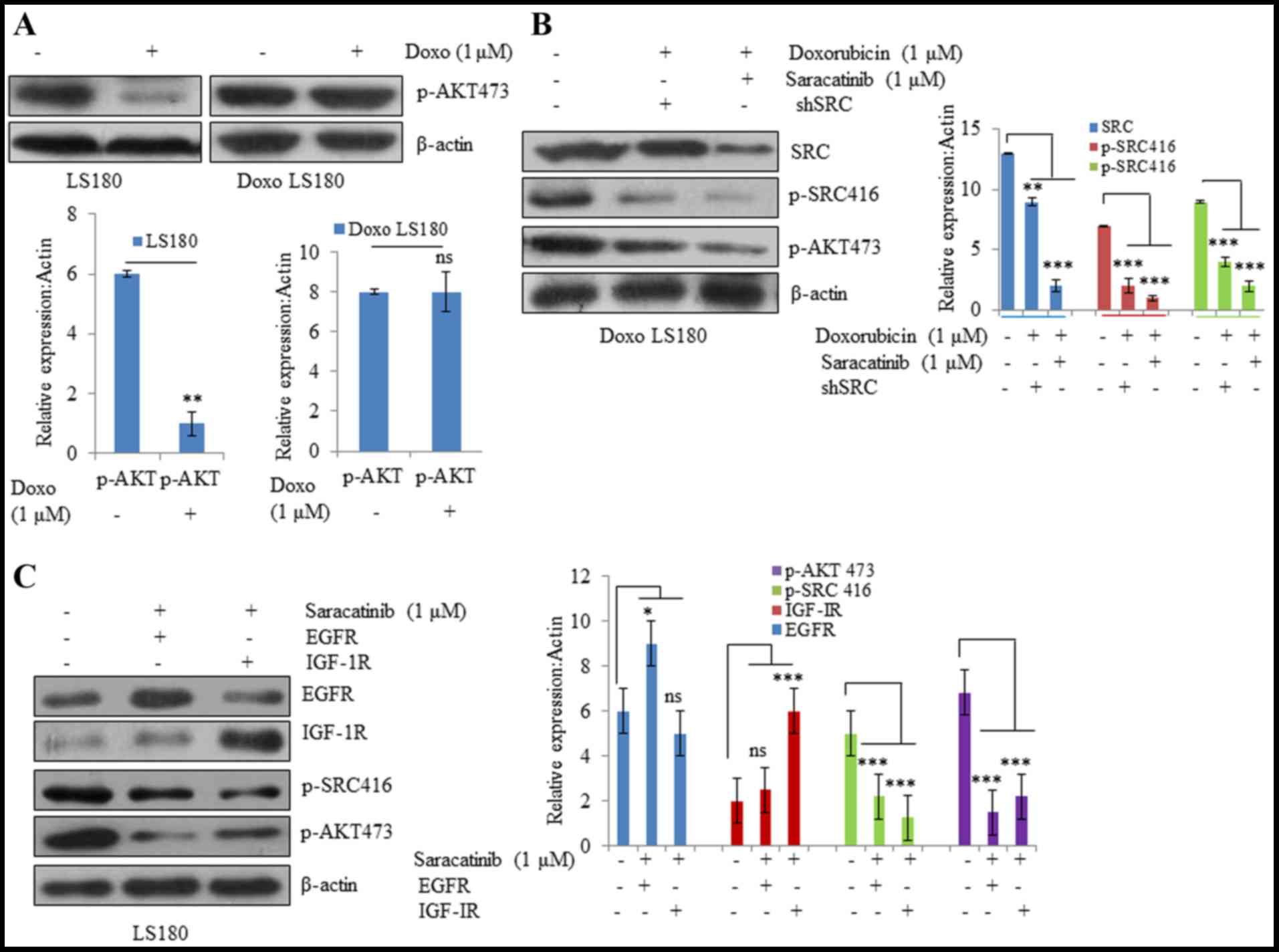 | Figure 4.SRC activation drives Doxo resistance.
(A) Parental LS180 cells responded to Doxo and there was a
significant decrease in phosphorylation of AKT; whereas in the
resistant cells, there was no significant difference in the p-AKT
levels following treatment with Doxo. (B) SRC inhibition by
saracatinib or SRC shRNA resensitized the resistant cells towards
Doxo and there was a decrease in the expression levels of p-AKT and
p-SRC. (C) SRC inhibition resulted in inhibition of AKT even in the
presence of overexpressed tyrosine kinases EGFR and IGF-1R. Data
are presented as the mean ± standard deviation of three different
experiments. Statistical comparisons were made using Bonferroni's
method. *P<0.05, **P<0.01 and ***P<0.001 vs. LS180 and
Doxo LS180 without Doxo treatment. SRC, steroid receptor
coactivator; Doxo, doxorubicin; Doxo LS180, doxorubicin-resistant
LS180 cells; p, phosphorylated; EGFR, epidermal growth factor
receptor; IGF-1R, insulin-like growth factor 1 receptor, ns, not
significant. |
Inhibiting SRC in vitro reverses
doxorubicin resistance
The present study demonstrated that SRC was driving
doxorubicin resistance and served as a common node of various
signaling pathways. Therefore, it was hypothesized that targeting
SRC may be effective in reverting doxorubicin resistance. In order
to test this hypothesis, the orally available SRC inhibitor,
saracatinib, was utilized. As demonstrated in Fig. 5, it was observed that saracatinib not
only sensitized the doxorubicin-resistant cells to doxorubicin, but
also induced inhibition of cell growth in EGFR- and
IGF-1R-overexpressing cells compared with doxorubicin (Fig. 5A). Additionally, 3D tumor spheroid
formation (Fig. 5D) and colony
forming potential (Fig. 5C) of
doxorubicin-resistant cells was inhibited when the cells were
treated with doxorubicin in combination with saracatinib.
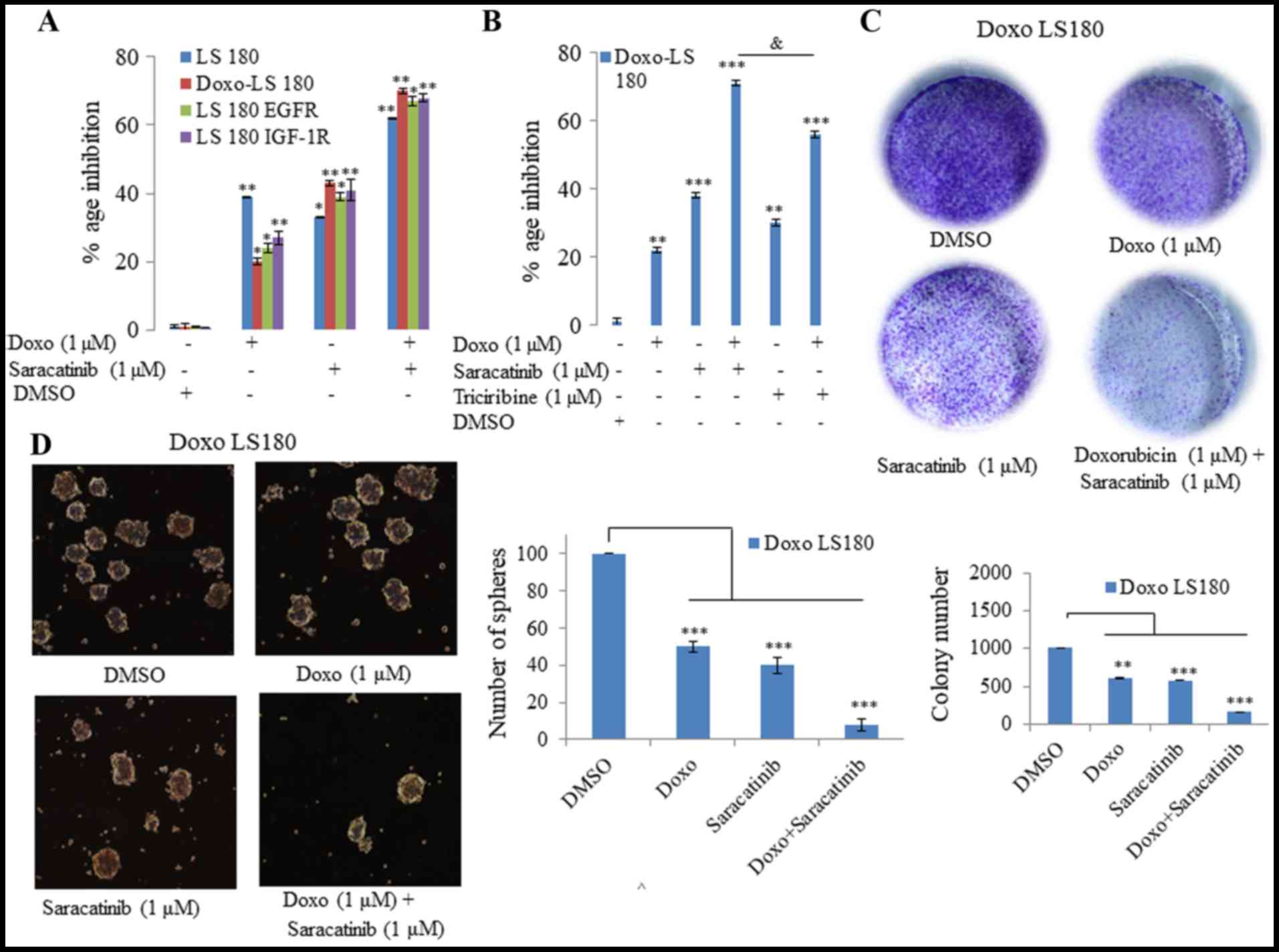 | Figure 5.SRC inhibition reverses Doxo
resistance in vitro. (A) Saracatinib in combination with
Doxo was more effective in all four models tested (LS180, Doxo
LS180, LS180 EGFR and LS180 IGF-1R) than when administered alone,
as determined by an MTT assay. (B) MTT assay determined that Doxo
in combination with the SRC inhibitor was more effective than a
combination of Doxo and AKT inhibitor, triciribine. (C) Combination
of SRC inhibitor, saracatinib, with Doxo inhibited the clonogenic
potential effectively in Doxo LS180 cells. (D) Doxo in combination
with SRC inhibitor, saracatinib, efficiently blocked the
proliferation of Doxo LS180 cells, as observed by inhibition of
mammospheres. Scale bar, 100 µM. Data are presented as the mean ±
standard deviation of three different experiments. Statistical
comparisons were made using Bonferroni's method. *P<0.05,
**P<0.01 and ***P<0.001 vs. LS180 and Doxo LS180 without Doxo
treatment. &P<0.05 as indicated. SRC, steroid
receptor coactivator; Doxo, doxorubicin; Doxo LS180,
doxorubicin-resistant LS180 cells; EGFR, epidermal growth factor
receptor; IGF-1R, insulin-like growth factor 1 receptor; DMSO,
dimethyl sulfoxide. |
Subsequently, the present study investigated whether
inhibition of AKT with its inhibitor, triciribine, induced the same
effect in combination with doxorubicin in doxorubicin-resistant
cells as that observed following SRC inhibition. However, it was
observed that AKT inhibition in combination with doxorubicin was
not as effective as SRC inhibition (Fig.
5B). Therefore, saracitinib demonstrated a greater effect at
re-sensitizing the cells towards doxorubicin than the AKT
inhibitor, triciribine.
Discussion
In colon cancer, which is one of the leading causes
of cancer-related mortality worldwide, drug resistance remains one
of the major challenges to be tackled (29). However, there are currently some
effective strategic approaches including the combination of
ATP-binding cassette transporter and EGFR inhibitors with
conventional drugs, against cancer that have been effective against
drug resistance in colon cancer (30). There are various targeted therapies
used for treatment of colon cancer, such as bevacizumab, which is a
Food and Drug Agency-approved first-line treatment, as well as a
second-line therapy for colon cancer approved in 2017 (31). Regorafenib was approved in 2012 for
metastatic colon cancer, and ramucirumab along with ziv-aflibercept
is the second-line treatment in metastatic colorectal cancer
(32). Doxorubicin, a potent
anticancer drug that is widely used to fight various cancer types,
including colon cancer, is cost effective compared with other
anticancer drugs (33). The present
study generated a drug-resistant cell line against doxorubicin by
treating the cell line with the drug for ~6 months. Drug resistance
induced EMT in the colon cancer cell line, LS180, as indicated by a
decrease in the expression of epithelial marker, E-cadherin, in
resistant cells as compared with the levels observed in parental
cells. There was a visible increase in the expression of
mesenchymal markers, including vimentin and N-cadherin, in the
drug-resistant cells compared with the expression in parental cells
at the mRNA and protein levels, as determined by RT-qPCR and
western blotting, respectively.
SRC, which acts as a signaling node between cell
surface receptors and cytoplasmic pathways, is also activated in
drug-resistant cells (34). In fact,
SRC activation appears to be the predominant factor responsible for
drug resistance, as it results in the activation of the downstream
AKT pathway (35). SRC is also
activated due to overexpression of RTKs, including EGFR and IGF-1R
(36,37). In the present study, it was
demonstrated that SRC inhibition not only resulted in reversal of
drug resistance, but also led to inactivation of AKT in resistant
cells. SRC was also inhibited upon inhibition of EGFR and IGF-1R.
The present results led to the conclusion that different RTKs,
along with their downstream targets that are activated due to drug
resistance, may be effectively blocked by SRC inhibition.
Furthermore, in vitro inhibition of SRC in combination with
doxorubicin also leads to inhibition of AKT in EGFR- and
IGF-1R-overexpressing cells. The present study revealed that 3D
tumor spheroid formation and colony forming potential of
doxorubicin-resistant cells was inhibited when cells were treated
with doxorubicin in combination with SRC inhibitor, saracatinib.
The present study also indicated that inhibition of AKT in
combination with doxorubicin was not as effective as treatment with
SRC inhibitor, saracatinib. Notably, previous studies have
suggested that paclitaxel and cisplatin inhibit SRC tyrosine
kinase, which enhances toxicity to human ovarian cancer cells as
well as mouse ovarian cancer cells (38,39).
In conclusion, the present study demonstrated that
SRC is activated in doxorubicin resistance, as well as
overexpression of RTKs, including EGFR and IGF-1R, which lead to
further activation of downstream pathways, such as the AKT
signaling pathway. Apoptosis assays in doxorubicin-resistant LS180
and parental LS180 cell lines are intended to be performed in
future studies. The combinatorial treatment of doxorubicin along
with inhibition of SRC not only helps in the inactivation of the
AKT pathway, but also in the reversal of resistance.
References
|
1
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sheth KR and Clary BM: Management of
hepatic metastases from colorectal cancer. Clin Colon Rectal Surg.
18:215–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aoyagi T, Terracina KP, Raza A and Takabe
K: Current treatment options for colon cancer peritoneal
carcinomatosis. World J Gastroenterol. 20:12493–12500. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nordlinger B, Sorbye H, Glimelius B,
Poston GJ, Schlag PM, Rougier P, Bechstein WO, Primrose JN, Walpole
ET, Finch-Jones M, et al: Perioperative chemotherapy with FOLFOX4
and surgery versus surgery alone for resectable liver metastases
from colorectal cancer (EORTC Intergroup trial 40983): A randomised
controlled trial. Lancet. 371:1007–1016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Colon Cancer Laparoscopic or Open
Resection Study Group. Buunen M, Veldkamp R, Hop WC, Kuhry E,
Jeekel J, Haglind E, Påhlman L, Cuesta MA, Msika S, et al: Survival
after laparoscopic surgery versus open surgery for colon cancer:
Long-term outcome of a randomised clinical trial. Lancet Oncol.
10:44–52. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diaz LA Jr, Williams RT, Wu J, Kinde I,
Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al:
The molecular evolution of acquired resistance to targeted EGFR
blockade in colorectal cancers. Nature. 486:537–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nikaido H: Prevention of drug access to
bacterial targets: Permeability barriers and active efflux.
Science. 264:382–388. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozben T: Mechanisms and strategies to
overcome multiple drug resistance in cancer. FEBS Lett.
580:2903–2909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ghannoum MA and Rice LB: Antifungal
agents: Mode of action, mechanisms of resistance, and correlation
of these mechanisms with bacterial resistance. Clin Microbiol Rev.
12:501–517. 1999.PubMed/NCBI
|
|
12
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim AY, Kwak JH, Je NK, Lee YH and Jung
YS: Epithelial-mesenchymal transition is associated with acquired
resistance to 5-fluorocuracil in HT-29 colon cancer cells. Toxicol
Res. 31:151–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosanò L, Cianfrocca R, Spinella F, Di
Castro V, Nicotra MR, Lucidi A, Ferrandina G, Natali PG and Bagnato
A: Acquisition of chemoresistance and EMT phenotype is linked with
activation of the endothelin A receptor pathway in ovarian
carcinoma cells. Clin Cancer Res. 17:2350–2360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Creighton CJ, Li X, Landis M, Dixon JM,
Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A,
Herschkowitz JI, et al: Residual breast cancers after conventional
therapy display mesenchymal as well as tumor-initiating features.
Proc Natl Acad Sci USA. 106:13820–13825. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hiscox S, Jiang WG, Obermeier K, Taylor K,
Morgan L, Burmi R, Barrow D and Nicholson RI: Tamoxifen resistance
in MCF7 cells promotes EMT-like behaviour and involves modulation
of beta-catenin phosphorylation. Int J Cancer. 118:290–301. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jones VS, Huang RY, Chen LP, Chen ZS, Fu L
and Huang RP: Cytokines in cancer drug resistance: Cues to new
therapeutic strategies. Biochim Biophys Acta. 1865:255–265.
2016.PubMed/NCBI
|
|
19
|
Sui H, Zhu L, Deng W and Li Q:
Epithelial-mesenchymal transition and drug resistance: Role,
molecular mechanisms, and therapeutic strategies. Oncol Res Treat.
37:584–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bromann PA, Korkaya H and Courtneidge SA:
The interplay between Src family kinases and receptor tyrosine
kinases. Oncogene. 23:7957–7968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guarino M: Src signaling in cancer
invasion. J Cell Physiol. 223:14–26. 2010.PubMed/NCBI
|
|
22
|
Alfarouk KO, Stock CM, Taylor S, Walsh M,
Muddathir AK, Verduzco D, Bashir AH, Mohammed OY, Elhassan GO,
Harguindey S, et al: Resistance to cancer chemotherapy: Failure in
drug response from ADME to P-gp. Cancer Cell Int. 15:712015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu C, Krishnan J and Xu XY: Intrinsic and
induced drug resistance mechanisms: In silico investigations at the
cellular and tissue scales. Integr Biol (Camb). 7:1044–1060. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Florea AM and Büsselberg D: Cisplatin as
an anti-tumor drug: Cellular mechanisms of activity, drug
resistance and induced side effects. Cancers (Basel). 3:1351–1371.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Imming P, Sinning C and Meyer A: Drugs,
their targets and the nature and number of drug targets. Nat Rev
Drug Discov. 5:821–834. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trédan O, Galmarini CM, Patel K and
Tannock IF: Drug resistance and the solid tumor microenvironment. J
Natl Cancer Inst. 99:1441–1454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jang M, Kim SS and Lee J: Cancer cell
metabolism: Implications for therapeutic targets. Exp Mol Med.
45:e452013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McDermott M, Eustace AJ, Busschots S,
Breen L, Crown J, Clynes M, O'Donovan N and Stordal B: In vitro
development of chemotherapy and targeted therapy drug-resistant
cancer cell lines: A practical guide with case studies. Front
Oncol. 4:402014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma X and Yu H: Global burden of cancer.
Yale J Biol Med. 79:85–94. 2006.PubMed/NCBI
|
|
31
|
Tartari F, Santoni M, Pistelli M and
Berardi R: Healthcare cost of HER2-positive and negative breast
tumors in the United States (2012–2035). Cancer treatment reviews.
60:12–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Giantonio BJ, Catalano PJ, Meropol NJ,
O'Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA and Benson AB III:
Eastern Cooperative Oncology Group Study E3200: Bevacizumab in
combination with oxaliplatin, fluorouracil, and leucovorin
(FOLFOX4) for previously treated metastatic colorectal cancer:
Results from the Eastern Cooperative Oncology Group Study E3200. J
Clin Oncol. 25:1539–1544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sonowal H, Pal PB, Wen JJ, Awasthi S,
Ramana KV and Srivastava SK: Aldose reductase inhibitor increases
doxorubicin-sensitivity of colon cancer cells and decreases
cardiotoxicity. Sci Rep. 7:31822017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Martino A, San KK, Daniel N and Ezekiel U:
Chemoresistance-induced epithelial-mesenchymal transition of a
colorectal cancer cell line. FASEB J. 29 Suppl 1:S721.172015.
|
|
35
|
Sen B and Johnson FM: Regulation of SRC
family kinases in human cancers. J Signal Transduct.
2011:8658192011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bromann PA, Korkaya H and Courtneidge SA:
The interplay between Src family kinases and receptor tyrosine
kinases. Oncogene. 23:7957–7968. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Furcht CM, Buonato JM and Lazzara MJ:
EGFR-activated Src family kinases maintain GAB1-SHP2 complexes
distal from EGFR. Sci Signal. 8:ra462015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pengetnze Y, Steed M, Roby KF, Terranova
PF and Taylor CC: Src tyrosine kinase promotes survival and
resistance to chemotherapeutics in a mouse ovarian cancer cell
line. Biochem Biophys Res Commun. 309:377–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen T, Pengetnze Y and Taylor CC: Src
inhibition enhances paclitaxel cytotoxicity in ovarian cancer cells
by caspase-9-independent activation of caspase-3. Mol Cancer Ther.
4:217–224. 2005.PubMed/NCBI
|