Introduction
Apoptosis, which is the best-described form of
programmed cell death, is a controlled and energy-dependent
process, the deregulation of which can lead to cancer (1). Over the past two decades, we have
witnessed explosive progress in this field. It is now known that
there are at least 3 pathways in apoptosis: The endogenous
mitochondrial pathway, the stress endoplasmic reticulum pathway and
the exogenous death receptor pathway (2). Among these, the role of the
mitochondrial pathway had been neglected for a long time. However,
researchers have found that mitochondria play an important role in
apoptosis by releasing key effector proteins from the mitochondrial
intermembrane space (IMS), including cytochrome c (Cyt-c)
and second mitochondria-derived activator of caspase (Smac)/DIABLO.
Mitochondrial outer membrane permeabilization (MOMP) results in the
release of IMS proteins, which supports the activation of
executioner caspases and is paramount for effective killing of
cancer cells (3,4).
Research has shown that the MOMP is the most
important point in apoptosis and that the mitochondrial pathway of
apoptosis is characterized by MOMP and activation of Cyt-c
(5). Moreover, the interaction
between the members of the Bcl-2 family can control the MOMP
(6). Bcl-2 family proteins are
subdivided into two groups on the basis of their pro- or
anti-apoptotic action. Anti-apoptotic family members include Bcl-2,
Bcl-xl, Bcl-w, Mcl-1 and A1/Bfl-1, while pro-apoptotic family
members include Bax, Bak and Bok/Mtd (7,8). Bcl-2
and Bcl-xl play anti-apoptotic roles by inhibiting their
pro-apoptotic counterparts and blocking the activation of caspase
in cytoplasm. The multidomain pro-apoptotic proteins, Bax and Bak,
are responsible for MOMP and are the master effectors of apoptosis
(9). In some cell types, the
extrinsic pathway can also cross with the mitochondrial pathway
through caspase-8 mediated cleavage of Bid, and the truncated tBid
will translocate to the mitochondria to trigger Cyt-c release
(10).
The expression and regulation of the Bcl-2 family
has a significant influence on the apoptosis. X-linked inhibitor of
apoptosis protein (XIAP) is the most potent caspase inhibitor of
the IAP family, whose inhibitory effects in the mitochondria during
apoptosis are well documented. In addition to its well-known
function in caspase suppression, XIAP can permeabilize and enter
mitochondria (11). XIAP comprises
three baculoviral IAP repeat domains (BIR1-3) and an interesting
zinc-finger RING domain, which exerts E3 ubiquitin ligase activity
(12). The XIAP E3 ligase activity
recruits endolysosomes into mitochondria, resulting in Smac
degradation. Through its E3 ligase, XIAP may possess more potential
biological functions.
So far, the mechanism by which XIAP regulates
mitochondrial function is not clear. Previous study has
demonstrated that XIAP serves as an E3 ligase for Bcl-2 and
stimulates UPS (Ubiquitin Proteasome System)-mediated degradation
of Bcl-2 (13). Since, there is a
close relationship between the Bcl-2 family and mitochondrial
function, we put forward the hypothesis that XIAP may impact
mitochondrial function during apoptosis by regulating the Bcl-2
family. We also investigated whether physiological or elevated XIAP
levels have a comprehensive effect on mitochondria.
Levels of XIAP were shown to be elevated in renal
cell carcinomas compared with normal renal cells. Previous research
has found that renal cell carcinomas are resistant to apoptosis
induced by chemical, immunological preparations and radiotherapy.
Moreover, a poor prognosis is associated with the over-expression
of XIAP in renal cell carcinomas (14). Therefore, we chose the Caki-1 cell
line, which over-express XIAP, and established a stable transfected
Caki-1 cell line with knockdown of XIAP using RNA interference.
Using these cells, we studied the dynamic change of the related
proteins in the process of apoptosis between the two cell
lines.
Materials and methods
Plasmids and molecular cloning
The XIAP sequence targeted by RNA interference in
this study was based on the data of Bilim et al (15), which verified that there were no
homologous sequences between this fragment and other gene sequences
and that the shRNA contained in this fragment can effectively
knockdown expression of the XIAP protein. The target sequence is
5′-AGGTGAAGGTGATAAAGTA-3′. The BLOCK-iT™ U6 RNAi Entry Vector kit
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was
used to generate the RNAi construct. Based on the selected target
sequence, the following oligonucleotide sequences for the shRNA
were used 5′-CACCGAGGTGAAGGTGATAAAGTACGAATACTTTATCACCTTCACC-3′ (top
strand DNA oligo);
5′-AAAAGGTGAAGGTGATAAAGTATTCGTACTTTATCACCTTCACCTC-3′ (bottom strand
DNA oligo).
These siRNA-encoding complementary single-stranded
oligonucleotides were hybridized to give Xho I-and Hind
III-compatible overhangs and then were ligated into pENTR™/U6. The
constructed plasmids were confirmed by sequencing and named
XIAP-shRNA-pENTR™/U6.
Cell culture and transfection
Caki-1 cells (renal carcinoma cell with
over-expression of XIAP) were purchased from China Infrastructure
of Cell Line Resources (Beijing, China). The cells were cultured in
minimum essential medium (MEM; Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Tianjin Kang Yuan Biological
Technology Co., Ltd., Tianjin, China). The cells were maintained in
a humidified 37°C incubator with 5% CO2. The cells were
split twice weekly. Cells in the logarithmic growth phase were used
for experiments.
Lipofectamine 2000 transfection reagent was used for
cell transfection (Gibco; Thermo Fisher Scientific, Inc.). The
BLOCK-iT™ U6 RNAi Entry Vector kit (Tianjin Kang Yuan Biological
Technology Co., Ltd.) was used to generate a construct to knock
down XIAP expression. Transfected cells were selected with G418 to
generate stable transfected clonal cells (Caki-1 cells successfully
transfected had G418 resistance) for 3–4 weeks. Stably transfected
Caki-1 cell clones were collected and named
Caki-1/XIAP-shRNA-pENTR™. with Caki-1/pENTR™ cells used as
controls. Because the PENTR™/U6 plasmid carries the GFP gene we
used a fluorescence microscope (AMG; Thermo Fisher Scientific,
Inc.) to identify transfected and no-transfected cells. After the
formation of the clonal cell lines, we picked a monoclonal cell
colony with green fluorescence and transferred it to a 24-well
plate for amplification.
Measurement of cell viability
Cell viability was determined by the MTT assay using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. We
used a MTT-kit (Gibco; Thermo Fisher Scientific, Inc.) to detect
the inhibition rate of cells exposed to different drug
concentrations. Etoposide was used to induce apoptosis.
Preparation of cell lysate and western
blotting
Total protein was extracted at different time points
after the induction of apoptosis. The extraction of cytoplasmic and
mitochondrial proteins was completed according to the Mitochondrial
Isolation kit instructions. Protein concentration was determined
using a bicinchoninic acid (BCA) protein assay kit. Whole cell
lysates were prepared using the RIPA reagent (Bioeasytech, Beijing,
China). A total of 40 µg of protein from each sample was resolved
by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to
polyvinylidene fluoride (PVDF) membranes. The membranes were
blocked with 5% non-fat milk overnight and probed with primary
antibodies against target proteins at 4°C overnight, followed by
incubation with a secondary anti-rabbit IgG-HRP antibody (1:10,000
dilution; Bioeasytech) or anti-mouse IgG-HRP antibody (1:20,000
dilution; Bioeasytech) at 37°C for 45 min. The following antibodies
were used: anti-XIAP, anti-apoptotic protease activating factor 1
(Apaf-1), anti-Smac, anti-Bcl-2, anti-Bax, anti-Bcl-xl (diluted
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-Cyt-c (diluted 1:500; Cell Signaling Technology, Inc.). Target
bands were visualized by enhanced chemiluminescence (ECL) solution
(WBKLS0500; EMD Millipore, Billerica, MA, USA) and analysed by
Gel-Pro-Analyzer software (Media. Cybernetics, Inc., Bethesda, MD,
USA). GAPDH was served as an internal control for cytoplasmic
samples, and COX IV was used as a loading control for mitochondrial
samples.
Statistical analysis
All values are reported as the mean ± standard
deviation, and differences between groups were analysed using a
two-tailed t-test or one-way analysis of variance (ANOVA) with the
Student-Newman-Keuls test, Statistical analysis was performed with
SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA).
Results
Effect of siRNA on protein expression
of XIAP gene
To reduce the expression of XIAP in Caki-1 cells,
sequence-specific siRNA of XIAP was synthesized, as mentioned in
the Materials and methods section. The transfected cells were
selected with for 3–4 weeks to generate stable transfected clones.
Three stable transfected cell clones were screened and one was
selected for further experiments. The interference efficiency of
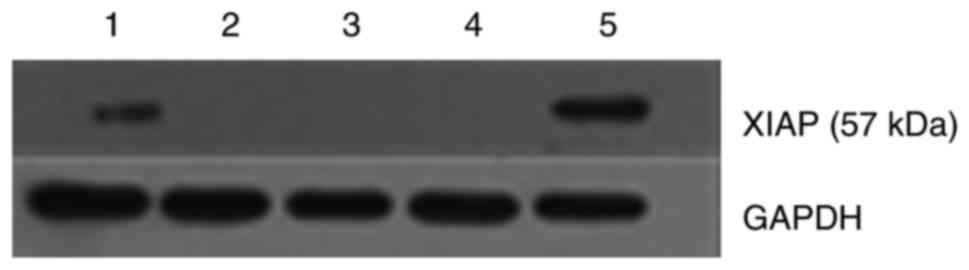
XIAP knockdown in cells was confirmed by western blot. As shown in
Fig. 1, XIAP shRNA transfection
markedly reduced the expression of XIAP, while there was no
significant difference between the Caki-1 control group and the
pENTR™/U6 group.
Knockdown of XIAP-sensitized Caki-1
cells to etoposide
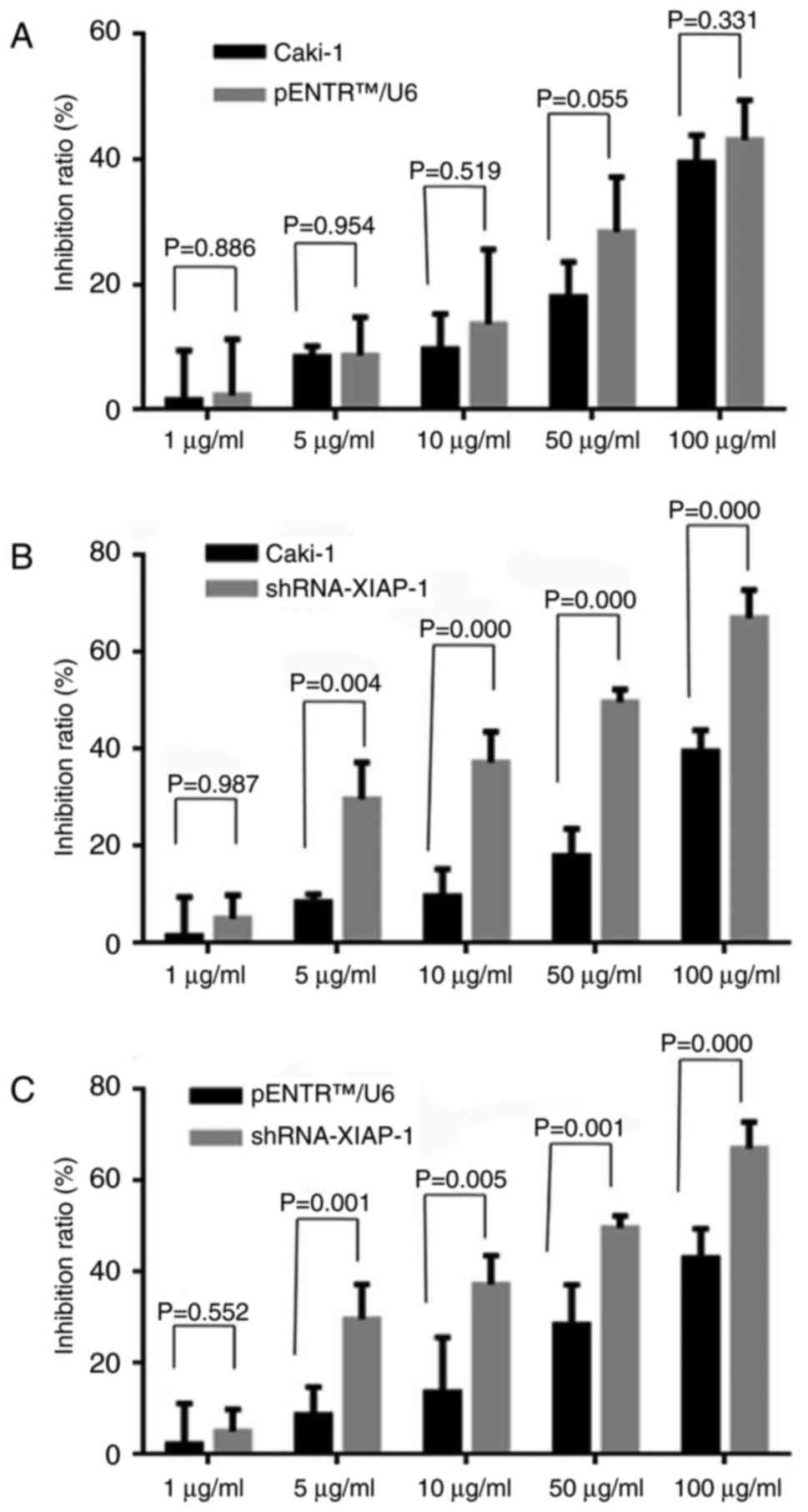
To evaluate whether the down-regulation of XIAP
expression would influence the anti-apoptotic ability of Caki-1
cells, the MTT method was used to detect the cell death rate under
different drug concentrations. As shown in Fig. 2, we observed that after knockdown of
XIAP protein expression, cell death rate was significantly
increased in response to different drug concentrations, while there
was no statistically significant difference between original Caki-1
cells and pENTR™U6 transfected Caki-1 cells.
XIAP impairs the release of
pro-apoptotic proteins from mitochondria
To comprehensively understand the effect of XIAP on
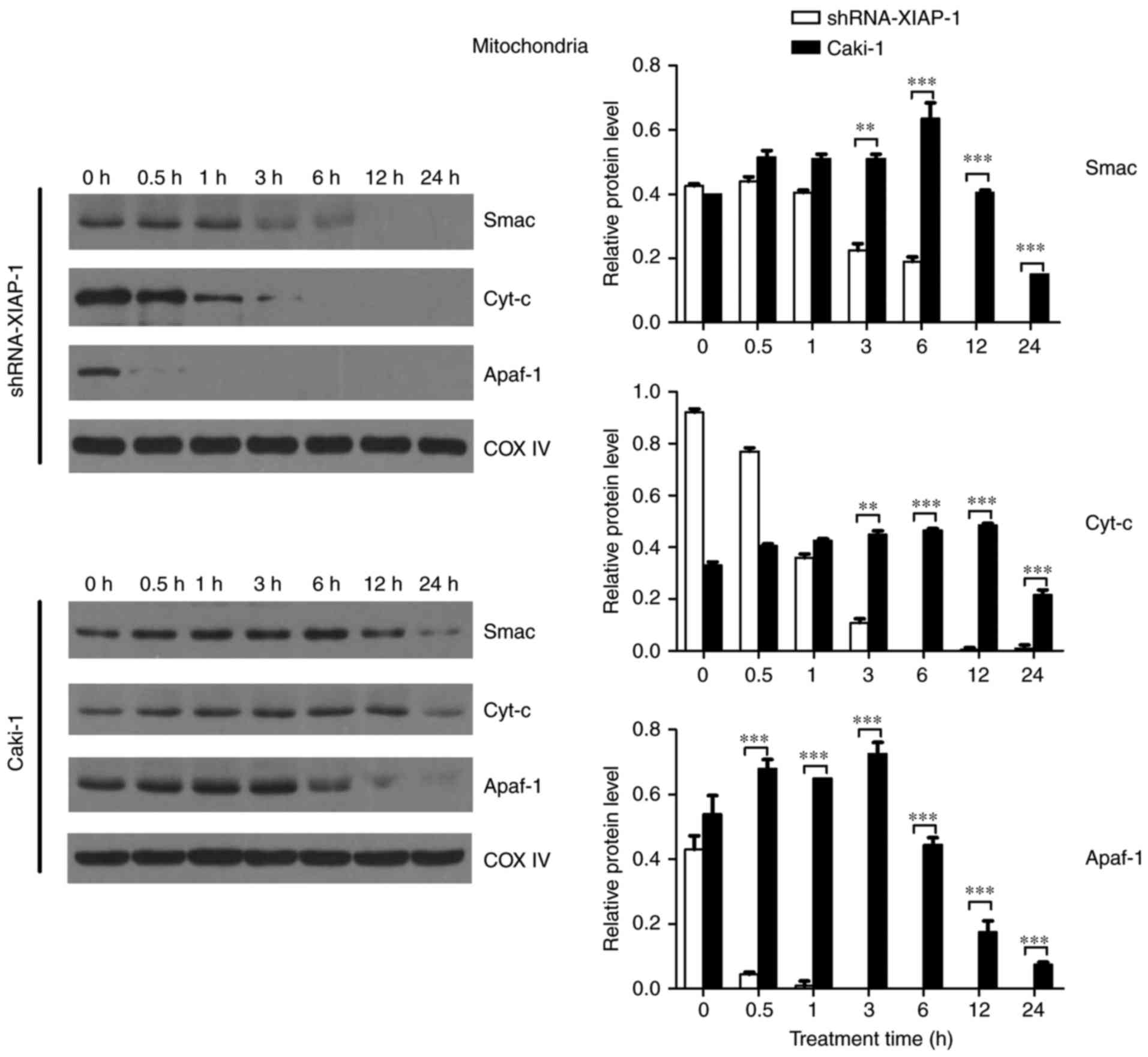
mitochondria, cells (Caki-1 and shRNA-XIAP-1) were treated with
etoposide for the indicated time intervals. At the end of etoposide
treatment, cytosolic and mitochondrial fractions were isolated, and
equal amounts of protein were subjected to western blotting to
detect Cyt-c, Smac, and Apaf-1). As shown in Fig. 3, comparing the dynamic changes of
proteins in the two cell lines, we found that the levels of
mitochondrial Cyt-c, Smac and Apaf-1 did not decrease rapidly in
response to etoposide in Caki-1 cells. Prior to 24 h, we did not
detect an obvious reduction in protein levels, indicating that the
release of pro-apoptotic proteins from mitochondria was delayed or
inhibited in response to the overexpression of XIAP in these renal
carcinoma cells. In contrast, in shRNA-XIAP-1-expressing cells
(knockdown of XIAP), the levels of mitochondrial Cyt-c, Smac and
Apaf-1 decreased rapidly after etoposide stimulation and by 6 h
after stimulation we could hardly detect these three proteins in
the mitochondria, meaning that after knockdown of XIAP in Caki-1
cells, the release time of pro-apoptotic proteins from mitochondria
was decreased relative to cells overexpressing XIAP. Additionally,
we found that Apaf-1 expression was decreased at 0.5 h, which was
more rapid than for the other proteins. We speculate that the
release mechanism of Apaf-1 is different from the other proteins.
The above results indicate that overexpression of XIAP impairs the
release of pro-apoptotic proteins from mitochondria and thereby
inhibits apoptosis. During apoptosis, Smac, which is similar to
Cyt-c, is released into the cytosol, and all pro-apoptotic proteins
play similar roles in the cytoplasm.
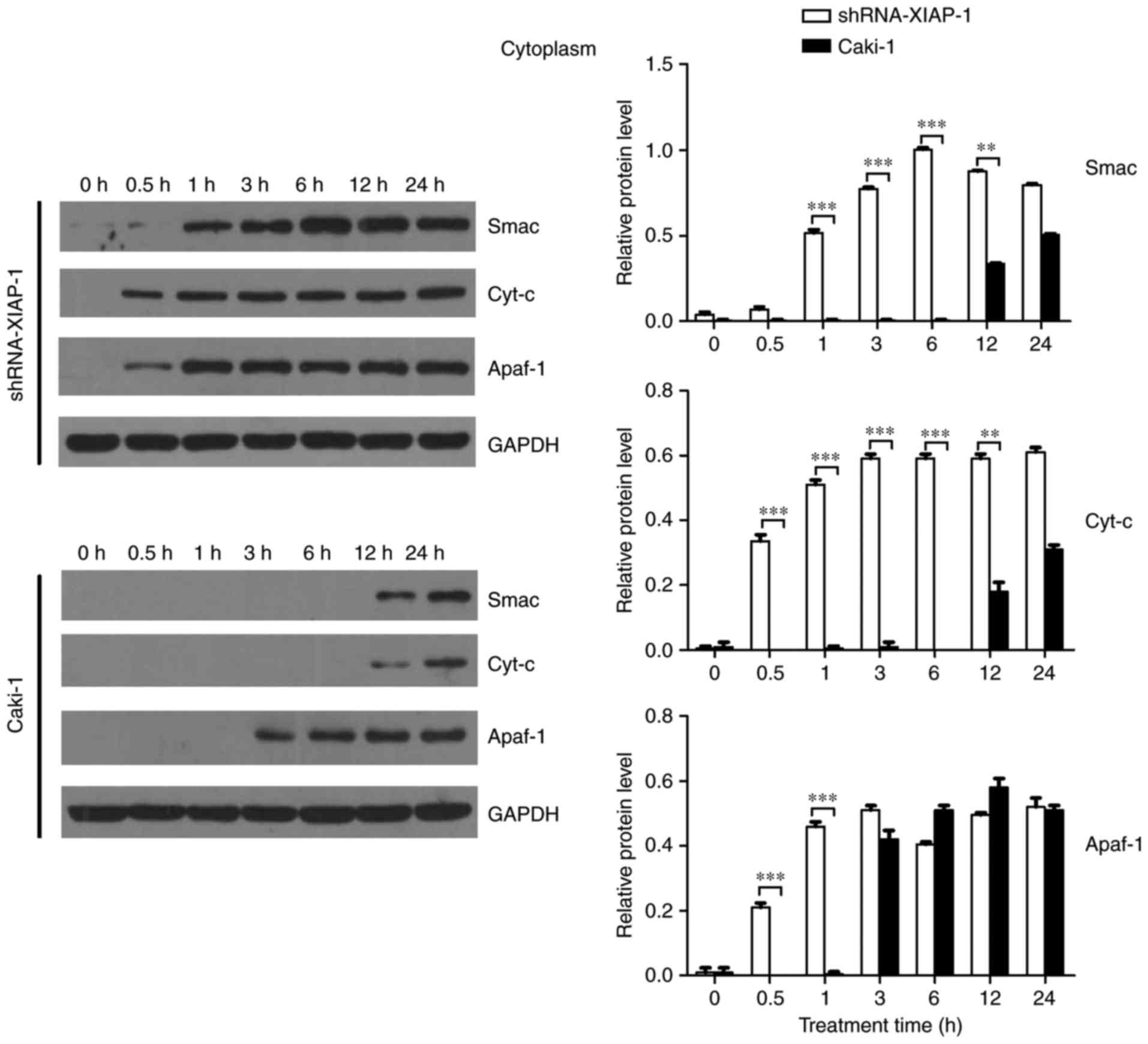
Therefore, we further confirmed the effect of XIAP
on mitochondria by detecting the dynamic changes of these proteins
in the cytoplasm. As shown in Fig. 4
and consistent with previous results, Cyt-c and Smac were not
detected in the cytoplasm until 12 h after apoptosis stimulation,
while Apaf-1 was detected in cytoplasm at 3 h after stimulation.
Upon knockdown of XIAP there was a noticeable difference with
pro-apoptotic proteins more rapidly increase after the stimulus,
with all three pro-apoptotic proteins being detected in cytoplasm
at 0.5 h. The time point of protein release from mitochondria was
obviously earlier than in cells overexpressing XIAP, supporting the
hypothesis that XIAP impairs the transfer of proteins from the
mitochondria to the cytoplasm. Moreover, the change in Smac
translocation was similar to that of Cyt-c. However, the trend for
Apaf-1 was different to that of Smac and Cyt-c. Thus, XIAP may
affect the release of Apaf-1 through a different mechanism.
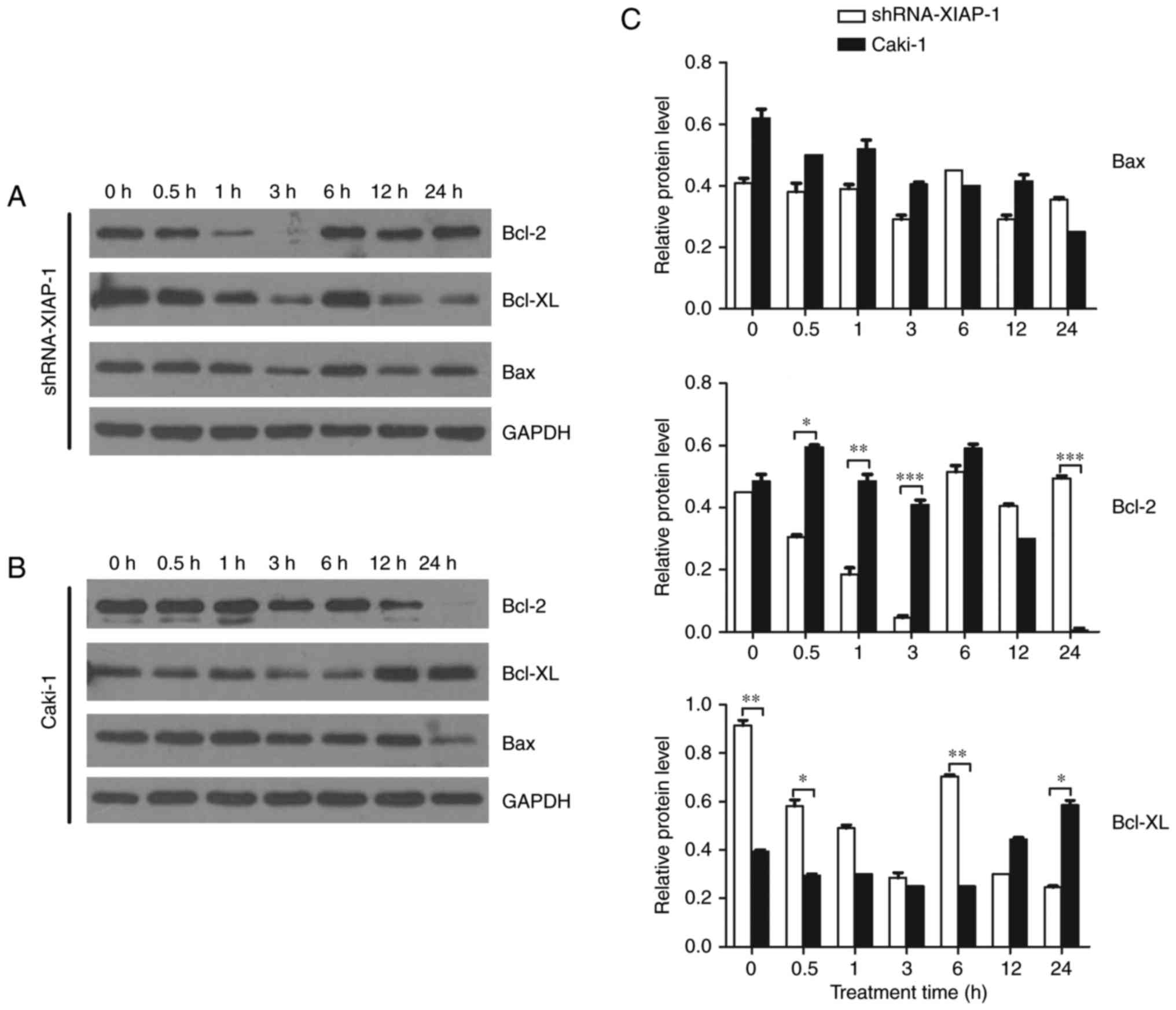
Bcl-2 family protein expression is
related to the XIAP protein
A number of studies have shown that the Bcl-2 family
plays an important role in the mitochondrial apoptosis pathway and
that the major site of action of the Bcl-2 family of proteins is on
the mitochondrial membrane. Therefore, we studied dynamic changes
in the Bcl-2 family proteins after the stimulation of apoptosis,
observing if there were differences between cell lines
overexpressing XIAP and cell lines with XIAP expression knocked
down. We selected Bcl-2 and Bcl-xl as our target pro-survival
proteins and Bax as our target pro-apoptotic protein. From the
results (Fig. 5), we can see that
after the stimulation of apoptosis, the pro-survival proteins,
Bcl-2 and Bcl-xl, were not significantly changed in Caki-1 cells.
However, in cells with XIAP expression knocked down, the level of
Bcl-2 and Bcl-xl decreased 10-fold and 4-fold, respectively at 3 h
after etoposide treatment, which indicates a decrease in the
restrainting effects upon Bax and Bak. For the pro-apoptotic
proteins, we found that there was no significant difference in Bax
expression in these two cell lines at each time point, indicating
that the function of Bax/Bak may only be regulated by a series of
conformational changes.
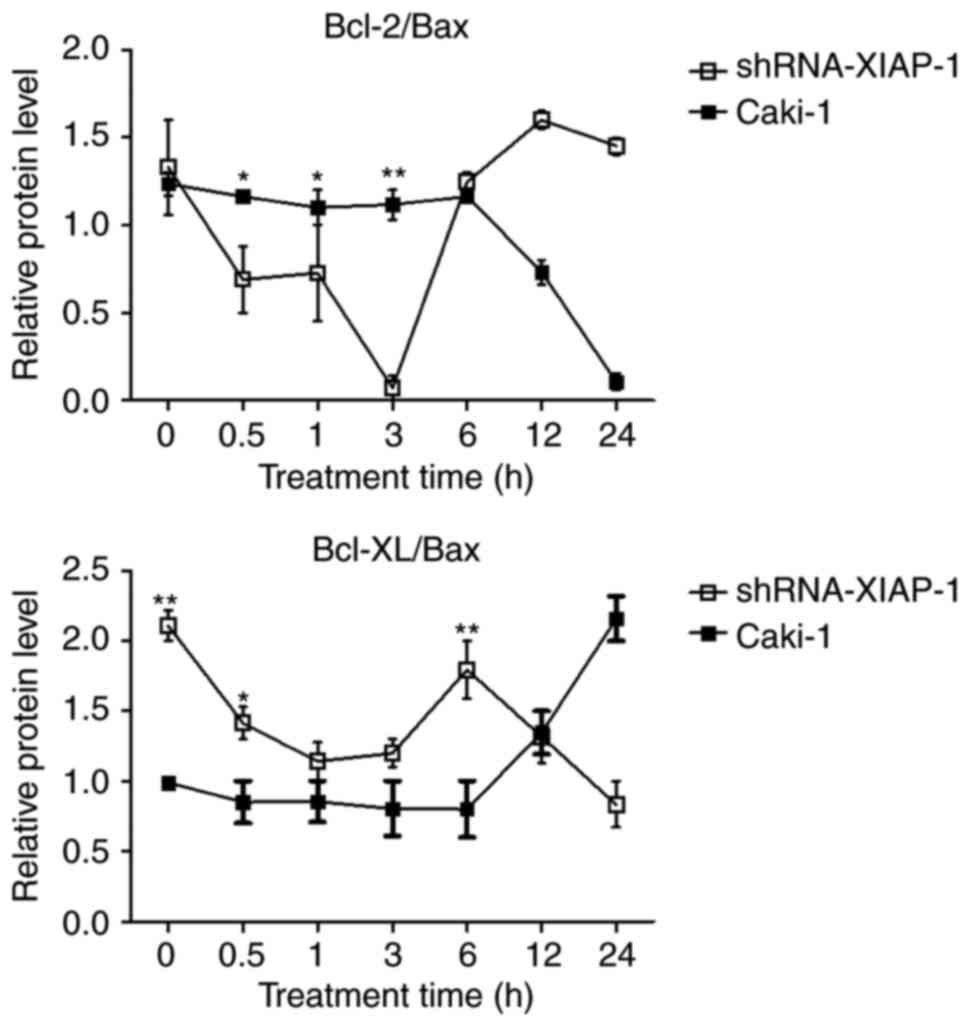
Imbalance of Bcl-2 family
proteins
Studies suggest that the mitochondrial apoptosis
pathway is regulated by balanced interactions between members of
the Bcl-2 family of proteins (16).
Therefore, we analysed the Bcl-2/Bax and Bcl-xl/Bax ratios
(Fig. 6). In Caki-1 cells, the
balance of Bcl-2/Bax and Bcl-xl/Bax were impaired at 12 h in
accordance with the time of release of pro-apoptotic proteins from
mitochondria, which was extended compared to the shRNA-XIAP-1
expressing cells. After knockdown of XIAP, the ratio was impaired
at 0.5 h after the stimulation of apoptosis, which was also in
accordance with the time of release of pro-apoptotic proteins from
mitochondria in shRNA-XIAP-1 expressing cells. From these results,
we can see that the change in XIAP expression impacts the balance
of Bcl-2 family proteins, which shows that XIAP has an important
impact upon the Bcl-2 family of proteins.
Discussion
Mitochondria play an important role in the process
of apoptosis, with Bcl-2 family proteins and Cyt-c closely
interacting with each other in the mitochondrial apoptotic pathway
(17). However, key issues regarding
upstream regulation of the Bcl-2 family remain largely unclear. In
addition to its well-known role in caspase suppression, XIAP can
localize to the mitochondria and activate Bax-mediated
mitochondrial permeabilization. Moreover, we previously reported
that XIAP can enter BH3-only protein-targeted or CCCP-depolarized
mitochondria, resulting in the degradation of its antagonist, Smac,
through endolysosomal and proteasomal actions (18). As MOMP-mediated release of Smac and
the consequent inhibition of XIAP are essential for activation of
apoptosis (19), this suggests an
additional pro-survival role for XIAP during apoptosis. Therefore,
we first studied the impact of XIAP on mitochondria. We then
observed changes in the Bcl-2 family of proteins to determine the
function and regulation of the Bcl-2 family by XIAP during
apoptosis signalling by regulating the level of XIAP through RNA
small interference.
The Bcl-2 family is a class of very conserved
proteins. Bcl-2 and Bcl-xL play a major role in the outer membrane
of the mitochondria by maintaining the integrity of the membrane.
They are the major anti-apoptotic proteins. Bax and Bak can destroy
the integrity of mitochondrial membrane, promoting the release of
Cyt-c and mediating apoptosis. They are the major pro-apoptotic
proteins (20). Therefore, we
selected Bcl-2, Bcl-xl and Bax as our targets to detect the effects
of altered XIAP expression.
Apoptosis is a rapid process. Therefore, it is very
meaningful to study the dynamics of protein-protein and
protein-membrane interactions in vivo as well as the
structures of membrane-bound proteins and captured conformational
changes, which are the key to understanding the mechanisms of both
apoptosis induction and execution. Therefore, in order to
comprehensively and dynamically evaluate the function of the Bcl-2
family proteins and mitochondria, we studied the dynamic changes in
pro-apoptotic proteins between the cytoplasmic and mitochondrial
compartments at different time points (at 0, 0.5, 1, 3, 6, 12 and
24 h) before and after the stimulation of apoptosis.
We showed that, compared with the knockdown of XIAP,
the presence of XIAP can prolong and limit the release of Cyt-c,
Smac and Apaf-1 from the mitochondria during etoposide-induced
apoptosis. The major goal of the mitochondrial apoptosis pathway is
the release of caspase activator (e.g., Cyt-c). The release of
Cyt-c occurs following a variety of death stimuli and has been
shown to activate Apaf-1, which in turn activates caspase-9 and
caspase-3 (21). The release of
these pro-apoptotic proteins is a marker of the mitochondrial
apoptosis pathway. Thus, the results of our studies clearly show
that XIAP affects the function of the mitochondria during
apoptosis. Additionally, we found that the changes in expression of
Apaf-1 are different from that of the other proteins, which
indicates that the release mechanism of Apaf-1 is different. This
difference in release mechanism of these proteins needs to be
studied more deeply. Another question is how XIAP affect
mitochondria. Thomas et al evaluated the apoptotic response
to drug treatment of B-CLL cells in vitro, finding that
cells with a high Bcl-2/Bax ratio were more drug resistant than
cells with a low Bcl-2/Bax ratio (22). In addition, researchers have shown
that the Bcl-2/Bax ratio correlates with ID50 values and clinical
responsiveness (23). Comparing the
ratio of Bcl-2/Bax and Bcl-xl/Bax in the two cell lines, our data
indicate that the level of Bcl-2 family members is primarily
mediated by XIAP and the balance of Bcl-2 family proteins is
related to XIAP. Thus, overexpression of XIAP influences changes in
Bcl-2 family members during the process of cell apoptosis, which
affect the function of mitochondria.
Taken together, our data show that the activity of
XIAP induced a previously unknown function at the mitochondria and
in the Bcl-2 family proteins. This study strengthens the
anti-apoptotic potential of XIAP and provides a new direction for
the treatment of cancer.
Acknowledgements
This study was supported by grants from National
Natural Science Foundation of China (no. 81441073) and Beijing
Municipal Commission of education science and technology plan
projects (no. KM201310025017).
References
|
1
|
Burz C, Berindan-Neagoe I, Balacescu O and
Irimie A: Apoptosis in cancer: Key molecular signaling pathways and
therapy targets. Acta Oncologica. 48:811–821. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jeong SY and Seol DW: The role of
mitochondria in apoptosis. BMB Rep. 41:11–22. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Newmeyer DD and Fergusonmiller S:
Mitochondria: Releasing power for life and unleashing the
machineries of death. Cell. 112:481–490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vo TT: Relative mitochondrial priming of
myeloblasts and normal HSCs determines chemotherapeutic success in
AML. Cell. 151:344–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chipuk JE and Green DR: How do BCL-2
proteins induce mitochondrial outer membrane permeabilization?
Trends Cell Biol. 18:157–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kvansakul M, Yang H, Fairlie WD, Czabotar
PE, Fischer SF, Perugini MA, Huang DC and Colman PM: Vaccinia virus
anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that
binds a highly selective subset of BH3-containing death ligands.
Cell Death Differ. 15:1564–1571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, Macgregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: A requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamacher-Brady A and Brady NR:
Bax/Bak-dependent, Drp1-independent Targeting of X-linked inhibitor
of apoptosis protein XIAP) into inner mitochondrial compartments
counteracts smac/DIABLO-dependent effector caspase activation. J
Biol Chem. 290:220052015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ,
Li P, Srinivasula SM, Alnemri ES, Fairman R and Shi Y: Mechanism of
XIAP-mediated inhibition of caspase-9. Mol Cell. 11:519–527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Edison N, Curtz Y, Paland N, Mamriev D,
Chorubczyk N, Haviv-Reingewertz T, Kfir N, Morgenstern D,
Kupervaser M, Kagan J, et al: Degradation of Bcl-2 by XIAP and ARTS
promotes apoptosis. Cell Rep. 21:442–454. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan Y, Mahotka C, Heikaus S, Shibata T,
Wethkamp N, Liebmann J, Suschek CV, Guo Y, Gabbert HE, Gerharz CD
and Ramp U: Disturbed balance of expression between XIAP and
Smac/DIABLO during tumour progression in renal cell carcinomas. Br
J Cancer. 91:1349–1357. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bilim V, Yuuki K, Itoi T, Muto A, Kato T,
Nagaoka A, Motoyama T and Tomita Y: Double inhibition of XIAP and
Bcl-2 axis is beneficial for retrieving sensitivity of renal cell
cancer to apoptosis. Br J Cancer. 98:941–949. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gavathiotis E, Reyna DE, Davis ML, Bird GH
and Walensky LD: BH3-triggered structural reorganization drives the
activation of proapoptotic BAX. Mol Cell. 40:481–492. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamacherbrady A, Choe SC, Krijnselocker J
and Brady NR: Intramitochondrial recruitment of endolysosomes
mediates Smac degradation and constitutes a novel intrinsic
apoptosis antagonizing function of XIAP E3 ligase. Cell Death
Differ. 21:1862–1876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jost PJ, Grabow S, Gray D, Mckenzie MD,
Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, et
al: XIAP discriminates between type I and type II FAS-induced
apoptosis. Nature. 460:1035–1039. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bossy-Wetzel E, Newmeyer DD and Green DR:
Mitochondrial cytochrome c release in apoptosis occurs upstream of
DEVD-specific caspase activation and independently of mitochondrial
transmembrane depolarization. EMBO J. 17:37–49. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thomas A, El Rouby S, Reed JC, Krajewski
S, Silber R, Potmesil M and Newcomb EW: Drug-induced apoptosis in
B-cell chronic lymphocytic leukemia: Relationship between p53 gene
mutation and bcl-2/bax proteins in drug resistance. Oncogene.
12:1055–1062. 1996.PubMed/NCBI
|
|
23
|
Pepper C, Hoy T and Bentley DP: Bcl-2/Bax
ratios in chronic lymphocytic leukaemia and their correlation with
in vitro apoptosis and clinical resistance. Br J Cancer.
76:935–938. 1997. View Article : Google Scholar : PubMed/NCBI
|