Introduction
Pneumonia is defined as the inflammation and
consolidation of lung tissue due to an infectious agent. It is a
leading cause of mortality worldwide, particularly in children and
the elderly (1,2). Typical symptoms of pneumonia include
fever, chills, pleuritic chest pain, and productive cough of
purulent sputum (1). Severe
pneumonia is defined as the admission to an intensive care unit
(ICU) and results in extremely high mortality rates (3). Chronic obstructive pulmonary disease
(COPD) is a major public health problem and is a leading cause of
chronic morbidity and mortality worldwide (4). COPD is particularly prevalent in the
elderly and it is a frequent comorbidity in patients with
community-acquired pneumonia (CAP) (5). During treatment with corticosteroids in
elderly patients with COPD, serious pneumonia typically occurs with
a high risk (6,7). The severity of COPD, including the
presence of pulmonary emphysema and the airflow limitation grade,
has been demonstrated to be independently associated with the
development of severe pneumonia (8).
Furthermore, CAP patients with COPD have a similar mortality rate
compared to those without COPD (9).
Therefore, it is necessary to elucidate the molecular mechanism
underlying severe pneumonia accompanied with COPD and compare it
with the mechanism underlying severe pneumonia.
Several molecular mechanisms associated with severe
pneumonia have been described. For instance, four risk
single-nucleotide polymorphisms located in chromosomes 1 and 17
have been identified as being significantly correlated with the
susceptibility to developing severe pneumonia in A/H1N1 infection
(10). Staphylococcal α-hemolysin is
an essential virulence factor in severe Staphylococcus
aureus pneumonia and α-hemolysin-mediated activation of the
NLRP3 inflammasome may induce necrotic pulmonary injury
(11). Furthermore, previous reports
have indicated that severe pneumonia is associated with
methicillin-resistant S. aureus carrying the staphylococcal
cassette chromosome mec type IV and Panton-Valentine leukocidin
genes (12,13). Furthermore, a recent study reported
that high expression of interleukin-10 (IL-10) and
interferon-induced protein (IP-10) in human immunodeficiency
virus-infected infants were associated with more severe hypoxic
pneumonia (14). The IL-6–174
GG genotype is correlated with lower severity and mortality in
patients with pneumococcal CAP (15). However, to date, there have been no
studies reporting the molecular mechanisms underlying severe
pneumonia accompanied with COPD, or comparing the similarities and
differences between patients with severe pneumonia alone and severe
pneumonia patients with COPD at the genetic level.
In the present study, mRNA-seq and bioinformatics
were used to analyze the differential expression of genes in the
peripheral blood from patients with severe pneumonia alone and
severe pneumonia accompanied with COPD. Genes that were
differentially expressed in the two groups of patients were
analyzed. These findings may provide novel information for the
understanding of the molecular mechanisms underlying severe
pneumonia and severe pneumonia accompanied with COPD.
Materials and methods
Clinical samples
This study was approved by the Medical Ethics
Committee of the Chinese People's Liberation Army (PLA) General
Hospital (Beijing, China). In total, 18 patients with pneumonia who
received therapy in Chinese PLA General Hospital between June 2013
and December 2013 were included in this study, including 9 patients
with severe pneumonia alone (the SP group), 9 patients with severe
pneumonia accompanied with COPD (the CSP group). Another 9
volunteers without pneumonia were enrolled as normal controls (the
C group). For inclusion, patients with severe pneumonia met at
least one of the following criteria: i) Disorders of consciousness;
ii) respiratory rate ≥30/min; iii) diastolic blood pressure <60
mmHg, PaO2/FiO2 <300, and mechanical
ventilation; iv) systolic blood pressure ≤90 mmHg; v) septic shock;
vi) bilateral or multilobar pneumonia by chest radiograph, or
lesions enlargement within 48 h after admission ≥50%; and vii)
oliguria: urine volume <20 ml/h or <80 ml/4 h, or acute renal
failure requiring dialysis treatment (16).
Peripheral blood samples were collected from each
patient and volunteer. Written informed consent was obtained before
sampling. In each group, three samples with an equal volume were
randomly pooled into one sample for sequencing. Therefore, three
sequencing samples were generated for each group: WLL1–3 for the SP
group, WLL4–6 for the CSP group, and WLL7-9 for the C group.
RNA extraction and Illumina
sequencing
Firstly, plasma was separated from the nine
sequencing samples, respectively. Total RNA was extracted and
purified from the plasma samples using an miRNeasy Serum/Plasma Kit
(Qiagen GmbH, Hilden, Germany). The quality control of RNA was
measured using the Agilent Bioanalyzer 2100 (Agilent Technologies,
Inc., Palo Alto, CA, USA). The bioanalyzer software automatically
generated the value of RNA Integrity Number (RIN, 1 to 10) based on
the ratio of the 18S to 28S ribosomal subunits to determine the
level of RNA degradation in gel electrophoresis, which removed
individual interpretation in RNA quality control. RNAs with RIN
≥8.0 were used in the study. Subsequently, poly (A) mRNA was
enriched by oligo (dT) magnetic beads (Dynabeads® oligo
dT, Dynal Inc., Great Neck, NY), and then interrupted into shot
fragments by fragmentation buffer (Agilent Technologies, Inc.,
Santa Clara, CA, USA). RNA fragments were then reverse transcribed
into cDNA. The concentration of cDNA in the library was quantified
into 1 ng/µl with a Qubit 2.0 fluorometer, and then the cDNA were
detected using the Agilent Bioanalyzer 2100 (Agilent Technologies,
Inc.). Libraries were pooled according to the data size and
effective cDNA concentration. Clusters of the cDNA libraries were
generated on an Illumina cBot system (Illumina, Inc., San Diego,
CA, USA). Finally, the cDNA libraries were sequenced using the
rapid mode 2×150 bp paired-end reads on an Illumina HiSeq™ 4000
system (Illumina, Inc.). Raw sequencing data was uploaded to the
public National Center for Biotechnology Information database under
the BioProject accession no. PRJNA325383.
Data filtering
Adapter sequences in the raw reads were removed
using Cutadapt (version 1.2.1; cutadapt.readthedocs.io/en/stable/)
(17), with at least 10-bp overlap
(AGATCGGAAG) and allowing for a 20% base error rate. Using a 5-bp
window, the mean quality value of each window was controlled to be
at least 20. Shortening of the 3′ terminal reads was permitted,
although the shortest length was at least 50 bp. Reads with unknown
sequences (‘N’) were removed.
Statistics and alignment of reads
The number of raw and clean reads, as well as the
percentage of clean reads, was summarized to ensure the validity
and reliability of the sequencing data. Furthermore, the index of
the reference genome was created using Bowtie 2 (version 2.1.0;
sourceforge.net/projects/bowtie-bio/files/bowtie2/2.1.0/),
and clean reads were aligned to the human genome (hg19) using
TopHat 2.1.1 software (ccb.jhu.edu/software/tophat/index.shtml).
Differential expression analysis of
genes
The read count of each gene was calculated by HTSeq
0.6.1p2 (huber.embl.de/users/anders/HTSeq) and subsequently
normalized using Trimmed Mean of M values normalization (18) in the Limma package (version 3.24.15;
bioconductor.org/packages/release/bioc/html/limma.html).
Normalized data were transformed to gene expression matrix using
the voom method (19).
Subsequently, differentially expressed genes (DEGs)
in the comparison groups of SP vs. C and CSP vs. C were identified
using the edgeR package of Bioconductor (bioconductor.org/packages/release/bioc/html/edgeR.html).
Only the genes meeting the criteria of |log2 FC (fold
change) |>1 and P<0.05 were identified as DEGs.
Functional analysis of the DEGs
Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analysis was performed for the identified DEGs
using the clusterProfiler (version 2.2.7; bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
package of R (20). The P-value of
each pathway term was calculated by Fisher's exact test (21), and only the pathway terms with
P<0.05 were considered significant.
Venn analysis of the DEGs
Gene symbols and their log2 FC values of
the identified DEGs were input into the VennPlex software (version
1.0.02; irp.nia.nih.gov/bioinformatics/vennplex.html)
(22) to compare the DEGs in the SP
and CSP groups. Common upregulated and downregulated genes between
the two groups were identified and defined as common DEGs.
Coexpression analysis of the common
DEGs
Pearson correlation coefficient (PCC) was used to
screen the coexpression pairs among all of DEGs. P-value was
calculated based on the Z-score of the coexpression pair, and |PCC|
>0.9 and P<0.05 were set as the cut-off criteria.
Furthermore, coexpression pairs of the common DEGs
were selected for the construction of the coexpression network.
Here, each pair must contain at least one common DEG. The network
was visualized by Cytoscape 3.3.0 (cytoscape.org/). In the network, a ‘node’ represents a
protein (or gene); a ‘line’ represents a coexpression pair of the
proteins. The ‘degree’ of each node is equal to the number of nodes
that coexpress with this node. The higher the degree is, the closer
the connections with other nodes are, indicating a more important
role of the node in the network.
Analysis of the potential
pneumonia-related genes
Comparative Toxicogenomics Database (CTD; ctdbase.org/) provides manually curated information
about chemical-gene/protein interactions, chemical-disease and
gene-disease relationships (23). In
this study, the keyword ‘pneumonia’ was used to search the genes
associated with pneumonia (April 26, 2016). The DEGs in the SP and
CSP groups were then compared with the pneumonia-related genes in
CTD using a venn tool (bioinformatics.lu/venn.php) in order to
identify the potential pneumonia-related genes from the DEGs.
Results
Data summary of quality control and
sequence alignment
Following quality control of the raw sequencing
data, the percentage of clean data was >97% in the nine samples
(Table I), indicating the sequencing
data as of high quality. The sequence alignment of the clean reads
to the hg19 indicated that overall mapping rates in the majority of
samples were between 60 and 75%. The concordant pair alignment rate
was 50–65% in the majority of samples (Table II).
 | Table I.Summary of the sequencing data after
quality control. |
Table I.
Summary of the sequencing data after
quality control.
| Sample | Raw data (Mb) | Clean data
(Mb) | Percent (%) |
|---|
| WLL1 | 15857.06 | 15457.13 | 97.48 |
| WLL2 | 30910.76 | 30140.40 | 97.51 |
| WLL3 | 14380.84 | 13989.76 | 97.28 |
| WLL7 | 16242.65 | 15811.34 | 97.34 |
| WLL8 | 17571.73 | 17114.66 | 97.40 |
| WLL9 | 12614.38 | 12283.53 | 97.38 |
| WLL4 | 12177.13 | 11909.79 | 97.80 |
| WLL5 | 21674.30 | 21182.59 | 97.73 |
| WLL6 | 19091.95 | 18678.84 | 97.84 |
| Table II.Data summary of the sequence
alignment. |
Table II.
Data summary of the sequence
alignment.
| Sample | Left mapped
reads | Right mapped
reads | Overall mapping
rate (%) | Concordant
alignment pair rate (%) |
|---|
| WLL1 | 40655723 | 31737852 | 68.50 | 53.20 |
| WLL2 | 68307024 | 48549459 | 56.70 | 39.30 |
| WLL3 | 36746057 | 28219712 | 67.80 | 52.80 |
| WLL4 | 31492239 | 27325889 | 72.50 | 62.10 |
| WLL5 | 57866494 | 49099958 | 74.00 | 62.30 |
| WLL6 | 50706071 | 42750383 | 73.40 | 61.40 |
| WLL7 | 38850721 | 28955703 | 62.60 | 46.50 |
| WLL8 | 45900593 | 36937197 | 70.70 | 57.80 |
| WLL9 | 32531013 | 25214426 | 68.70 | 55.00 |
Identification of DEGs
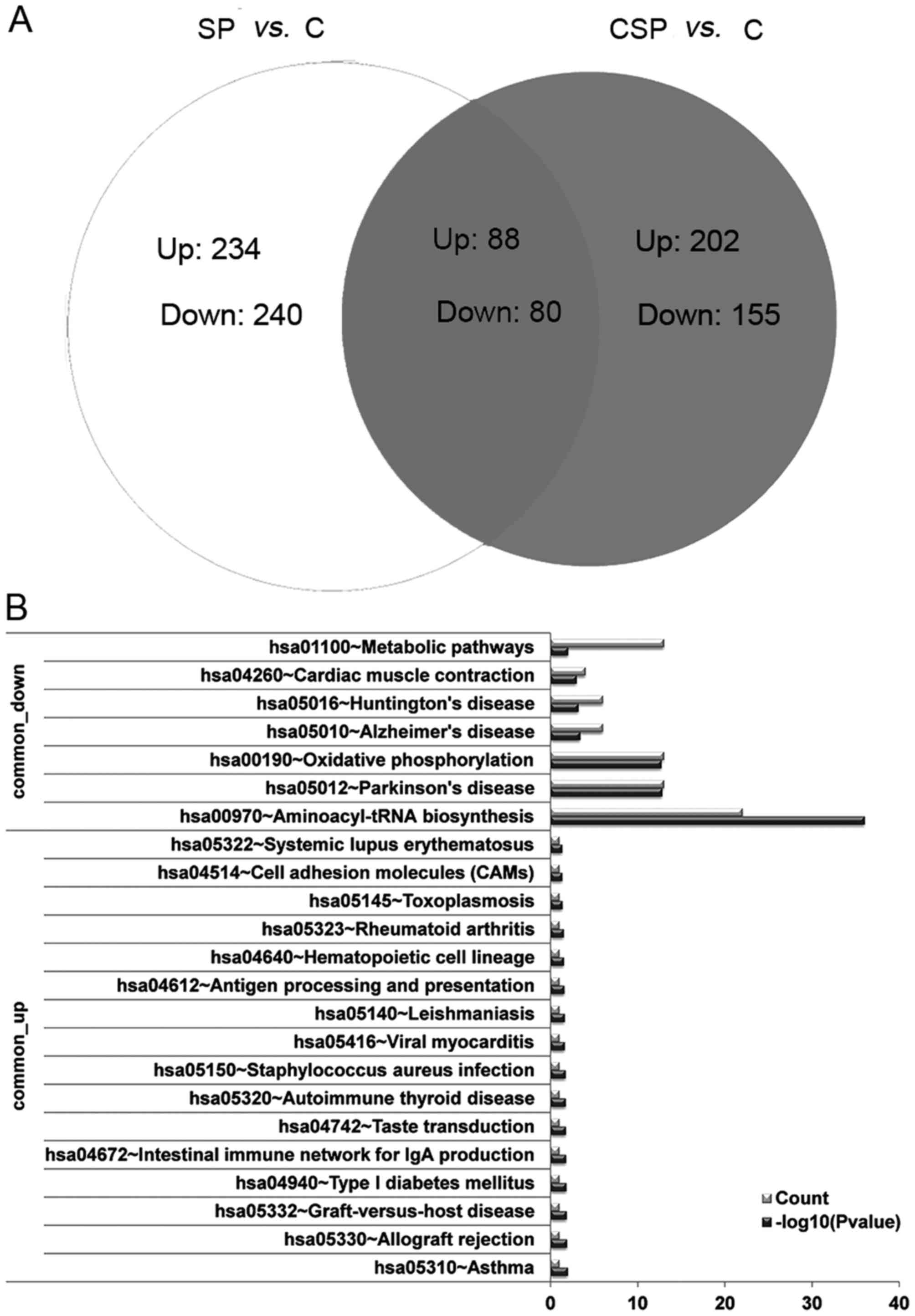
Based on the criteria, a total of 645 DEGs (323
upregulated and 322 downregulated genes) were identified in the SP
group, along with 528 DEGs (292 upregulated and 236 downregulated
genes) in the CSP group, compared with the C group. These DEGs were
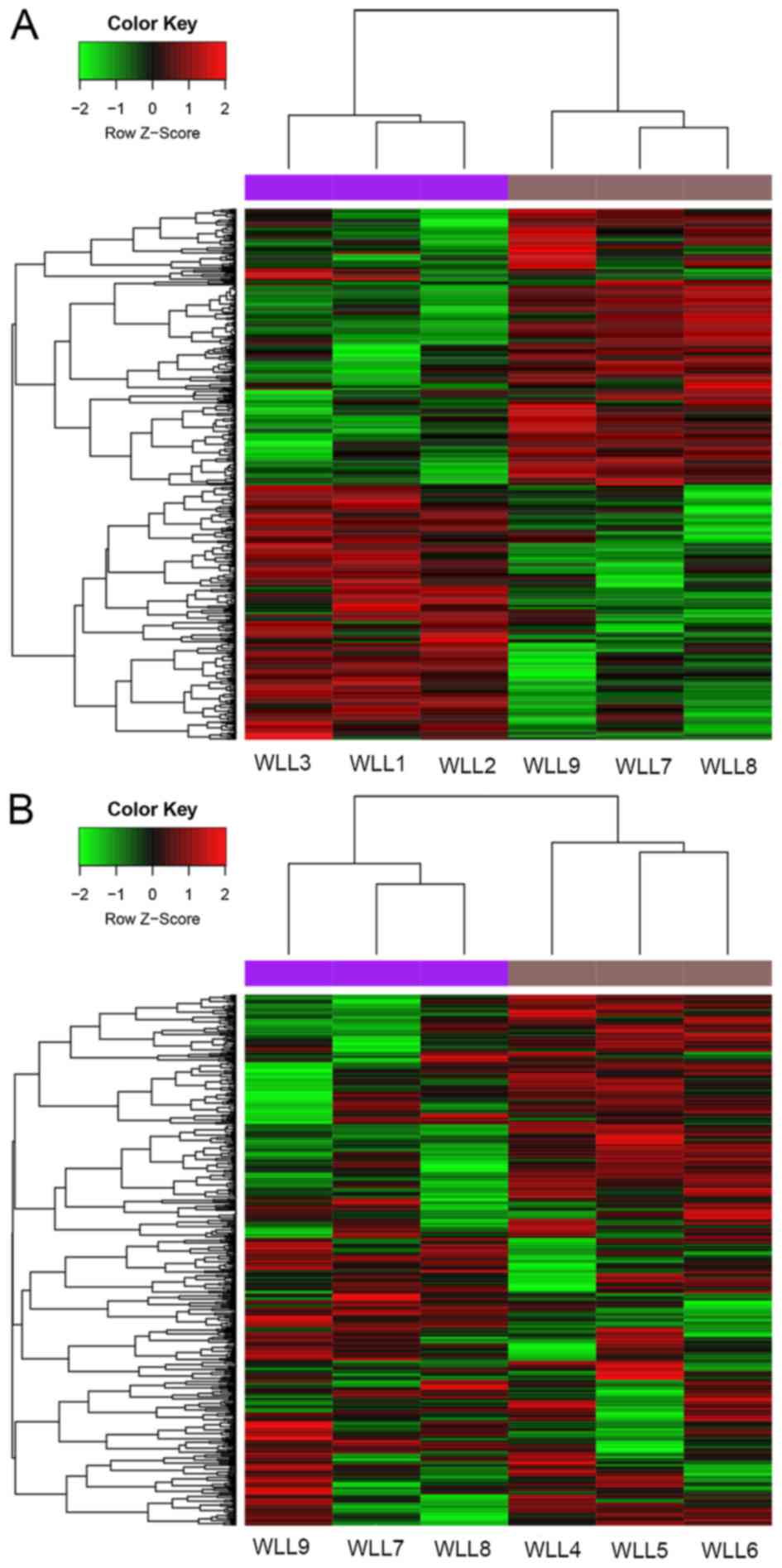
able to distinguish the two group samples (Fig. 1A and B).
Pathway enrichment analysis of
DEGs
To further investigate the potential biological
functions of the identified DEGs, KEGG pathway enrichment analysis
was performed. For the DEGs in the SP group, the upregulated gene
HLA-DRB5 was significantly enriched in the pathways
associated with asthma, allograft rejection and intestinal immune
network for IgA production, which were related to immunity. The
downregulated genes were mainly enriched in the pathways associated
with aminoacyl-tRNA biosynthesis (TRNS1 and TRNM) and
oxidative phosphorylation (COX3 and ND4L; Table III).
| Table III.Enriched pathway terms of
differentially expressed genes in the patients with severe
pneumonia alone compared with the controls. |
Table III.
Enriched pathway terms of
differentially expressed genes in the patients with severe
pneumonia alone compared with the controls.
| Pathway ID | Description | P-value | Genes |
|---|
| Upregulated |
|
hsa05310 | Asthma | 3.12E-02 | HLA-DRB5 |
|
hsa05330 | Allograft
rejection | 3.91E-02 | HLA-DRB5 |
|
hsa05332 | Graft-versus-host
disease | 4.30E-02 | HLA-DRB5 |
|
hsa04940 | Type I diabetes
mellitus | 4.50E-02 | HLA-DRB5 |
|
hsa04672 | Intestinal immune
network for IgA production | 4.89E-02 | HLA-DRB5 |
| Downregulated |
|
hsa00970 | Aminoacyl-tRNA
biosynthesis | 1.18E-30 | TRNS1, TRNM, TRNW,
TRNG, TRNH, TRNL1, TRNN, |
| | | TRNC, TRNA, TRNP,
TRNE, TRNT, TRNV, TRNS2, TRND, |
| | | TRNI, TRNQ, TRNK,
TRNY, TRNL2, TRNF, TRNR |
|
hsa05012 | Parkinson's
disease | 1.35E-10 | COX3, ND4L, COX2,
ND1, COX1, ND4, ATP8, |
| | | CYTB, ND3, ND5,
ND2, ATP6, ND6 |
|
hsa00190 | Oxidative
phosphorylation | 1.64E-10 | COX3, ND4L, COX2,
ND1, COX1, ND4, ATP8, |
| | | CYTB, ND3, ND5,
ND2, ATP6, ND6 |
|
hsa05016 | Huntington's
disease | 1.59E-03 | GPX1, COX3, COX2,
COX1, ATP8, CYTB, ATP6 |
|
hsa05010 | Alzheimer's
disease | 4.89E-03 | COX3, COX2, COX1,
ATP8, CYTB, ATP6 |
|
hsa04260 | Cardiac muscle
contraction | 5.92E-03 | COX3, COX2, COX1,
CYTB |
Furthermore, for the DEGs in the CSP group, the
upregulated genes (such as UGT2B17) were markedly enriched
in the pathways of metabolism, including starch and sucrose
metabolism, and ascorbate and aldarate metabolism. The
downregulated genes were significantly enriched in the
aminoacyl-tRNA biosynthesis (TRNS1 and TRNM) and
oxidative phosphorylation (COX3 and ND4L) pathways
(Table IV), which is similar to
those of the downregulated genes in the SP group.
| Table IV.Enriched pathway terms of
differentially expressed genes in the severe pneumonia patients
accompanied with chronic obstructive pulmonary disease compared
with the controls. |
Table IV.
Enriched pathway terms of
differentially expressed genes in the severe pneumonia patients
accompanied with chronic obstructive pulmonary disease compared
with the controls.
| Pathway ID | Description | P-value | Genes |
|---|
| Upregulated |
|
hsa00500 | Starch and sucrose
metabolism | 2.23E-03 | UGT2B17, AMY1C |
|
hsa00980 | Metabolism of
xenobiotics by cytochrome P450 | 3.82E-03 | UGT2B17, GSTM1 |
|
hsa00982 | Drug
metabolism-cytochrome P450 | 4.04E-03 | UGT2B17, GSTM1 |
|
hsa00053 | Ascorbate and
aldarate metabolism | 3.48E-02 | UGT2B17 |
|
hsa05310 | Asthma | 4.13E-02 | HLA-DRB5 |
|
hsa00040 | Pentose and
glucuronate interconversions | 4.26E-02 | UGT2B17 |
| Downregulated |
|
hsa00970 | Aminoacyl-tRNA
biosynthesis | 5.87E-36 | TRNL1, TRNS2, TRNR,
TRNV, TRNH, TRNC, |
| | | TRNN, TRNP, TRNY,
TRNT, TRNG, TRNE, TRNQ, |
| | | TRNI, TRNF, TRNS1,
TRNW, TRNA, TRND, |
| | | TRNK, TRNL2,
TRNM |
|
hsa05012 | Parkinson's
disease | 3.63E-13 | COX1, COX2, COX3,
ND1, ATP6, ND4L, ND4, |
| | | ATP8, ND3, ND2,
CYTB, ND5, ND6 |
|
hsa00190 | Oxidative
phosphorylation | 4.43E-13 | COX1, COX2, COX3,
ND1, ATP6, ND4L, ND4, |
| | | ATP8, ND3, ND2,
CYTB, ND5, ND6 |
|
hsa05010 | Alzheimer's
disease | 5.47E-04 | COX1, COX2, COX3,
ATP6, ATP8, CYTB |
|
hsa05016 | Huntington's
disease | 8.60E-04 | COX1, COX2, COX3,
ATP6, ATP8, CYTB |
|
hsa04260 | Cardiac muscle
contraction | 1.28E-03 | COX1, COX2, COX3,
CYTB |
|
hsa01100 | Metabolic
pathways | 1.63E-02 | COX1, COX2, COX3,
ND1, ATP6, ND4L, ND4, |
| | | ATP8, ND3, ND2,
CYTB, ND5, ND6 |
Venn analysis of the DEGs and the
functions of the common DEGs
To further investigate whether there are genes that
are differentially expressed in the two pneumonia groups, the DEGs
in the SP and CSP groups were compared. According to the venn
diagram, 88 DEGs were upregulated in the two groups (named
common-upregulated genes) and 80 were downregulated in the two
groups (named common-downregulated genes; Fig. 2A).
Furthermore, pathway enrichment analysis of the
common DEGs revealed that the commonly upregulated gene,
HLA-DRB5, was significantly enriched in the pathways
associated with asthma, allograft rejection and intestinal immune
network for IgA production. The commonly downregulated genes were
enriched in the pathways aminoacyl-tRNA biosynthesis (TRNS1
and TRNM) and oxidative phosphorylation (COX3 and
ND4L) pathways (Fig. 2B). The
functions of the common DEGs were similar to those of the DEGs in
the SP group.
Analysis of the coexpression
network
In total, 805 coexpression pairs of common genes
were chosen to construct a network. The coexpression network
consisted of 245 nodes and 805 coexpression pairs. A series of 69
commonly upregulated genes and 65 commonly downregulated genes were
included in the network (Fig.
3).
Some genes had a higher degree in the network,
including ND3 (degree, 36), ND4L (degree, 36),
TRNT (degree, 35), ND6 (degree, 35), TRNP
(degree, 35), ATP8 (degree, 35), ND1 (degree, 35),
MTND2P28 (degree, 35) and MTATP6P1 (degree, 34). All
of these genes were the commonly downregulated genes. Among the
commonly upregulated genes, several genes exhibited high degrees,
including TSPY6P (degree, 15), CDY10P (degree, 12),
TTTY15 (degree, 11), RNA5SP233 (degree, 11),
OFD1P3Y (degree, 9) and RN7SL255P (degree, 8;
Fig. 3).
Identification of potential
pneumonia-related genes
Based on the information in CTD, 131 DEGs were
identified to be associated with pneumonia, including 77 DEGs in
the SP group and 54 DEGs in the CSP group (Table V). Among these DEGs, 4 commonly
upregulated genes (MIR449A, TAS2R43, MIR656, and
EIF5AP4) and 16 commonly downregulated genes (NOXO1,
COX1, COX2, COX3, ND4, ND1, ND2, ND6, CYTB, ND3, SH3BGRL, ND5,
ATP6, CPXCR1, VCX3B, and CT45A3) were included.
| Table V.Potential pneumonia-related genes
identified from the differentially expressed genes based on the
Comparative Toxicogenomics Database. |
Table V.
Potential pneumonia-related genes
identified from the differentially expressed genes based on the
Comparative Toxicogenomics Database.
| Category | Potential
pneumonia-related genes |
|---|
| Severe pneumonia
alone group | PF4, TIMP1, GPX1,
B2M, FTH1, NOXO1, PPBP, SDPR, TMSB4X, COX1, PCBP1, |
| SH3BGRL3, NRGN,
COX2, LRRC26, RGS18, FLNA, ITGB3, TCEAL7, SAA4, ACOT1, |
| HAPLN4, COX3, ND4,
HBA1, HLA-E, HIST1H2AH, ND1, PGRMC1, TUBB, ND2, |
| RPL15, MIR148B,
ND6, MIR205, CYTB, APOBEC3B, ND3, PCDH20, MIR449A, |
| SH3BGRL, HLA-DRB5,
USP17L9P, ND5, MIR193A, HBA2, SNORA12, MIR559, |
| MIR572, TAS2R43,
CSNK2B, SPANXB1, SLX1A, FTHL17, LY6G6E, SLC10A3, |
| MIR339, RNR1,
MIR513B, HLA-DRB6, MIR4300, MIR656, ATP6, POTEF, CPXCR1, |
| RN7SK, FAM138A,
RPS18P9, SNORD115-30, MIR873, MIR190A, RBMY2EP, RNU1-1, |
| EIF5AP4, VCX3B,
MIR4499, CT45A3 |
| Severe pneumonia
accompanied with chronic obstructive pulmonary disease group | GSTM1, JUND, NOXO1,
COX1, COX2, COX3, MRPL12, ND4, ND1, SPANXD, ND2, |
|
| RPS20, ND6, CYTB,
MDP1, ND3, EIF4EBP3, MIR93, HNRNPU, MIR449A, MIR100, |
|
| SH3BGRL, RPS4Y2,
HLA-DRB5, IGKV1-17, ND5, UGT2B17, TAS2R43, MT1P3, |
| POM121L8P, MIR188,
LCE3C, GLOD5, CYB5D1, HBG1, MED14OS, INSL5, MIR656, |
| SNORD113-3, ATP6,
DTX2P1-UPK3BP1-PMS2P11, CPXCR1, DUX4L4, SNORA28, |
| MIR190A, F8A3,
MIR19B2, OR7E125P, EIF5AP4, MIR216A, VCX3B, TRBV21-1, |
| RNU4-2, CT45A3 |
Discussion
In the present study, based on the mRNA-seq and
bioinformatics findings, a total of 645 and 528 DEGs were
identified in the patients with severe pneumonia alone and severe
pneumonia patients with COPD, respectively, compared with the
normal controls. Among these DEGs, 88 upregulated genes and 80
downregulated genes were common between the two groups. In the
coexpression network, the commonly downregulated genes (including
ND1, ND3, ND4L, and ND6) and the commonly upregulated
genes (including TSPY6P and CDY10P) had a higher
degree. In addition, 131 DEGs (including COX1, COX2, COX3, ND1,
ND3, and ND6) were predicted to be potential
pneumonia-related genes.
ND1, ND3, ND4L and ND6 encode the
subunits of NADH dehydrogenase, which participate in the
mitochondrial oxidative phosphorylation, functioning in the
transfer of electrons from NADH to the respiratory chain (24). A previous study has demonstrated that
the type I NADH dehydrogenase of the human pathogen
Mycobacterium tuberculosis (Mtb) is able to neutralize
NOX2-derived reactive oxygen species to inhibit tumor
necrosis factor-α-mediated host macrophage apoptosis in innate
immunity (25). Differentially
expressed NADH dehydrogenase subunits were detected during the
immune response of Ostrea edulis against bonamiosis
(26). These results indicate the
association of NADH dehydrogenase with immunity, which is an
essential factor in the progression of pneumonia. In the present
study, ND1, ND3 and ND6 were identified as potential
pneumonia-related genes. Therefore, these genes encoding NADH
dehydrogenase subunits may have crucial roles in the progression of
severe pneumonia.
Commonly downregulated genes, COX1, COX2 and
COX3, were also predicted as potential pneumonia-related
genes. These three genes all encode isoforms of cyclooxygenase,
which has a critical role in various normal physiological functions
and regulates a variety of pathological conditions (27). Previous studies have demonstrated the
associations of COX2 with inflammation and immunity
(27,28). In the immune response of pneumonia,
COX2 expression is induced in lung lesions (27,29,30),
which is inconsistent with the result of the current study. This
disparity may be due to the difference of sample types used for
COX2 detection. In our future studies, COX2
expression will be determined in lung tissue and peripheral blood
of patients with severe pneumonia and normal control subjects.
With the exception of the downregulated genes,
several commonly upregulated genes also had a higher degree in the
coexpression network, including TSPY6P and CDY10P,
which are pseudogenes. In recent years, the biological functions of
pseudogenes have been increasingly investigated, and pseudogenes
have been reported to serve a role in formation of tumors (31). However, to the best of our knowledge,
there have been no reports on the functions of TSPY6P and
CDY10P. Therefore, the present study was the first to
demonstrate TSPY6P and CDY10P were deregulated in
patients with severe pneumonia, and were thus worthy of further
research.
Furthermore, in this study, 4 commonly upregulated
genes (MIR449A, TAS2R43, MIR656 and EIF5AP4) were
identified as potential pneumonia-related genes. MIR449A and
MIR656 encode miR-449a and miR-656, respectively. A previous
study has demonstrated that miRNA-449a modulates the expression of
the inflammatory marker, YKL40, by targeting components of
the NOTCH signaling pathway, indicating the important role of
miR-449a in the inflammatory response (32). TAS2R43 encodes taste 2
receptor member 43, which belongs to the large TAS2R receptor
family. An agonist for TAS2R1 and other TAS2Rs, amarogentin,
is able to decrease IL-8 and MMP-1 expression in mast
cells (33), suggesting the
potential association of TAS2R43 with immunity. Therefore,
MIR449A and TAS2R43 may have a critical role in the
progression of severe pneumonia, possibly via participating in
inflammation and immunity. However, MIR656 and
EIF5AP4 have not yet been reported to be related to
pneumonia, thus, they are novel genes that are potentially
associated with severe pneumonia.
Although a set of upregulated and downregulated
genes were common in the samples from patients with severe
pneumonia alone and patients with severe pneumonia accompanied with
COPD, there were a series of genes that were differentially
expressed between the two patient groups according to the venn
diagram analysis performed in the present study. The differences in
molecular mechanisms between severe pneumonia and severe pneumonia
accompanied with COPD will be further investigated in our future
studies.
In conclusion, the downregulated genes encoding NADH
dehydrogenase subunits (ND1, ND3, ND4L and ND6) and
the upregulated genes (TSPY6P, CDY10P, MIR449A, TAS2R43,
MIR656, and EIF5AP4) are newly-identified genes that may
be associated with the progression of severe pneumonia. These genes
will be further researched in our future studies. These results
provide novel information for further experimental studies to build
upon and contribute to the continued understanding of the molecular
mechanisms underlying severe pneumonia.
Acknowledgements
The present study was supported by grants from the
Welfare Industry Research Program of Ministry of Health (grant nos.
201502019), the National Natural Science Fund (grant nos. 81272060,
81371561, 81701961), the Youth Training Program of the PLA (grant
no. 16QNP135), Beijing Scientific And Technologic Supernova
Supportive Project (grant no. Z15111000030000/XXJH2015B100), the
PLA General Hospital Science and Technology Innovation Nursery Fund
Project (grant no. 16KMM56, 2017FC-WJFWZX-30) and the PLA Logistic
Major Science And Technology Project (grant nos. 14CXZ005,
AWS15J004 and BWS14J041).
References
|
1
|
Marrie TJ: Community-acquired pneumonia.
Clin Infect Dis. 18:501–515. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas CP, Ryan M, Chapman JD, Stason WB,
Tompkins CP, Suaya JA, Polsky D, Mannino DM and Shepard DS:
Incidence and cost of pneumonia in Medicare beneficiaries. Chest.
142:973–981. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ewig S, Ruiz M, Mensa J, Marcos MA,
Martinez JA, Arancibia F, Niederman MS and Torres A: Severe
community-acquired pneumonia: Assessment of severity criteria. Am J
Respir Crit Care Med. 158:1102–1108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nazir SA and Erbland ML: Chronic
obstructive pulmonary disease. Drugs Aging. 26:813–831. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iyer Parameswaran G and Murphy TF: Chronic
obstructive pulmonary disease: Role of bacteria and updated guide
to antibacterial selection in the older patient. Drugs Aging.
26:985–995. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calverley PM, Anderson JA, Celli B,
Ferguson GT, Jenkins C, Jones PW, Yates JC and Vestbo J: TORCH
investigators: Salmeterol and fluticasone propionate and survival
in chronic obstructive pulmonary disease. N Engl J Med.
356:775–789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh S, Amin AV and Loke YK: Long-term
use of inhaled corticosteroids and the risk of pneumonia in chronic
obstructive pulmonary disease: A meta-analysis. Arch Intern Med.
169:219–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eom JS, Song WJ, Yoo H, Jeong BH, Lee HY,
Koh WJ, Jeon K and Park HY: Chronic obstructive pulmonary disease
severity is associated with severe pneumonia. Ann Thorac Med.
10:105–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liapikou A, Polverino E, Ewig S, Cillóniz
C, Marcos MA, Mensa J, Bello S, Martin-Loeches I, Menéndez R and
Torres A: Severity and outcomes of hospitalised community-acquired
pneumonia in COPD patients. Eur Respir J. 39:855–861. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zuniga J, Buendía-Roldán I, Zhao Y,
Jiménez L, Torres D, Romo J, Ramírez G, Cruz A, Vargas-Alarcon G,
Sheu CC, et al: Genetic variants associated with severe pneumonia
in A/H1N1 influenza infection. Eur Respir J. 39:604–610. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kebaier C, Chamberland RR, Allen IC, Gao
X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL and Duncan
JA: Staphylococcus aureus α-hemolysin mediates virulence in a
murine model of severe pneumonia through activation of the NLRP3
inflammasome. J Infect Dis. 205:807–817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gillet Y, Vanhems P, Lina G, Bes M,
Vandenesch F, Floret D and Etienne J: Factors predicting mortality
in necrotizing community-acquired pneumonia caused by
Staphylococcus aureus containing Panton-Valentine leukocidin. Clin
Infect Dis. 45:315–321. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kilic A, Li H, Stratton CW and Tang YW:
Antimicrobial susceptibility patterns and staphylococcal cassette
chromosome mec types of, as well as Panton-Valentine leukocidin
occurrence among, methicillin-resistant Staphylococcus aureus
isolates from children and adults in middle Tennessee. J Clin
Microbiol. 44:4436–4440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Green R, Terclanche A, Becker P, Rheeder
P, Wittenberg DF, Anderson R and Masekela R: Cytokine profile and
clinical correlates in HIV-exposed infants with severe (hypoxic)
pneumonia. South African Respirat J. 22:3–6. 2016. View Article : Google Scholar
|
|
15
|
Martín-Loeches I, Solé-Violán J, de Castro
Rodríguez F, García-Laorden MI, Borderías L, Blanquer J, Rajas O,
Briones ML, Aspa J, Herrera-Ramos E, et al: Variants at the
promoter of the interleukin-6 gene are associated with severity and
outcome of pneumococcal community-acquired pneumonia. Intensive
Care Med. 38:256–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Association RdbotCm, . The diagnosis and
treatment guideline of community-acquired pneumonia. Chin J Tuberc
Respir Dis. 29:651–655. 2006.(In Chinese).
|
|
17
|
Martin M: Cutadapt removes adapter
sequences from high-throughput sequencing reads. EMBnet J.
17:10–12. 2011. View Article : Google Scholar
|
|
18
|
Robinson MD and Oshlack A: A scaling
normalization method for differential expression analysis of
RNA-seq data. Genome Biol. 11:R252010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Law CW, Chen Y, Shi W and Smyth GK: Voom:
Precision weights unlock linear model analysis tools for RNA-seq
read counts. Genome Biol. 15:R292014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Routledge R: Fisher's Exact Test.
Encyclopedia of Biostatistics. 3:2005. View Article : Google Scholar
|
|
22
|
Cai H, Chen H, Yi T, Daimon CM, Boyle JP,
Peers C, Maudsley S and Martin B: VennPlex-a novel Venn diagram
program for comparing and visualizing datasets with differentially
regulated datapoints. PLoS One. 8:e533882013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Davis AP, Grondin CJ, Lennon-Hopkins K,
Saraceni-Richards C, Sciaky D, King BL, Wiegers TC and Mattingly
CJ: The comparative toxicogenomics database's 10th year
anniversary: Update 2015. Nucleic Acids Res. 43:D914–D920. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sparks LM, Xie H, Koza RA, Mynatt R,
Hulver MW, Bray GA and Smith SR: A high-fat diet coordinately
downregulates genes required for mitochondrial oxidative
phosphorylation in skeletal muscle. Diabetes. 54:1926–1933. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miller JL, Velmurugan K, Cowan MJ and
Briken V: The type I NADH dehydrogenase of Mycobacterium
tuberculosis counters phagosomal NOX2 activity to inhibit
TNF-α-mediated host cell apoptosis. PLoS Pathog. 6:e10008642010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martín-Gómez L, Villalba A and Abollo E:
Identification and expression of immune genes in the flat oyster
Ostrea edulis in response to bonamiosis. Gene. 492:81–93. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Khan KN, Stanfield K, Trajkovic D and
Harris RK: Cyclooxygenase-2 expression in inflammatory lung lesions
of nonhuman primates. Vet Pathol. 37:512–516. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zaiss AK, Zuber J, Chu C, Machado HB, Jiao
J, Catapang AB, Ishikawa TO, Gil JS, Lowe SW and Herschman HR:
Reversible suppression of cyclooxygenase 2 (COX-2) expression in
vivo by inducible RNA interference. PLoS One. 9:e1012632014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sadikot RT, Zeng H, Azim AC, Joo M, Dey
SK, Breyer RM, Peebles RS, Blackwell TS and Christman JW: Bacterial
clearance of Pseudomonas aeruginosa is enhanced by the inhibition
of COX-2. Eur J Immunol. 37:1001–1009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giacominelli-Stuffler R, Marruchella G,
Storelli M, Sabatucci A, Angelucci CB and Maccarrone M:
5-Lipoxygenase and cyclooxygenase-2 in the lungs of pigs naturally
affected by enzootic pneumonia and porcine pleuropneumonia. Res Vet
Sci. 93:898–903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Poliseno L, Salmena L, Zhang J, Carver B,
Haveman WJ and Pandolfi PP: A coding-independent function of gene
and pseudogene mRNAs regulates tumour biology. Nature.
465:1033–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarma NJ, Tiriveedhi V, Subramanian V,
Shenoy S, Crippin JS, Chapman WC and Mohanakumar T: Hepatitis C
virus mediated changes in miRNA-449a modulates inflammatory
biomarker YKL40 through components of the NOTCH signaling pathway.
PLoS One. 7:e508262012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wölfle U, Haarhaus B and Schempp CM:
Amarogentin displays immunomodulatory effects in human mast cells
and keratinocytes. Mediators inflamm. 2015:6301282015. View Article : Google Scholar : PubMed/NCBI
|