Introduction
Infantile neuroaxonal dystrophy (INAD), also known
as Seitelberger's disease (Online Mendelian of Inheritance in Man
ID 256600), is a rare autosomal recessive neurodegenerative
disorder. Classic INAD manifests as psychomotor regression from the
age of 6 months to 3 years, followed by tetraparesis resulting from
neurologic deterioration, cerebellar ataxia, optic atrophy and
dementia (1). INAD is caused by
mutation of the phospholipase A2 group VI (PLA2G6) gene, mapped to
chromosome 22q13.1 (2,3). The present case report described the
clinical presentation and genetic analysis of an 18-month-old
female patient with INAD caused by PLA2G6 mutations with an unusual
phenotype.
Materials and methods
Case report
An 18-month-old female patient of non-consanguineous
healthy Chinese parents without significant family history
presented at the First Affiliated Hospital of Anhui Medical
University (Hefei, China) in October 2015. The patient was born at
40 weeks of gestation after a normal pregnancy and delivery. Her
weight was 3.3 kg and her head circumference was 33 cm at birth.
The Apgar scores (4) were 10 at 1
and 5 min and the total bilirubin serum levels were 252.76 µmol/l 3
days following birth. No feeding difficulties or hypotonia were
noticed. Her motion and mental development were normal at 8 months
after birth. At the age of 2 months, she achieved head control. Her
parents noticed the onset of nystagmus at 6 months. She recognized
her parents and attempted to call for them. When the patient was 8
months old, she achieved independent sitting. However, at the age
of 14 months, the patient was not able to stand independently and
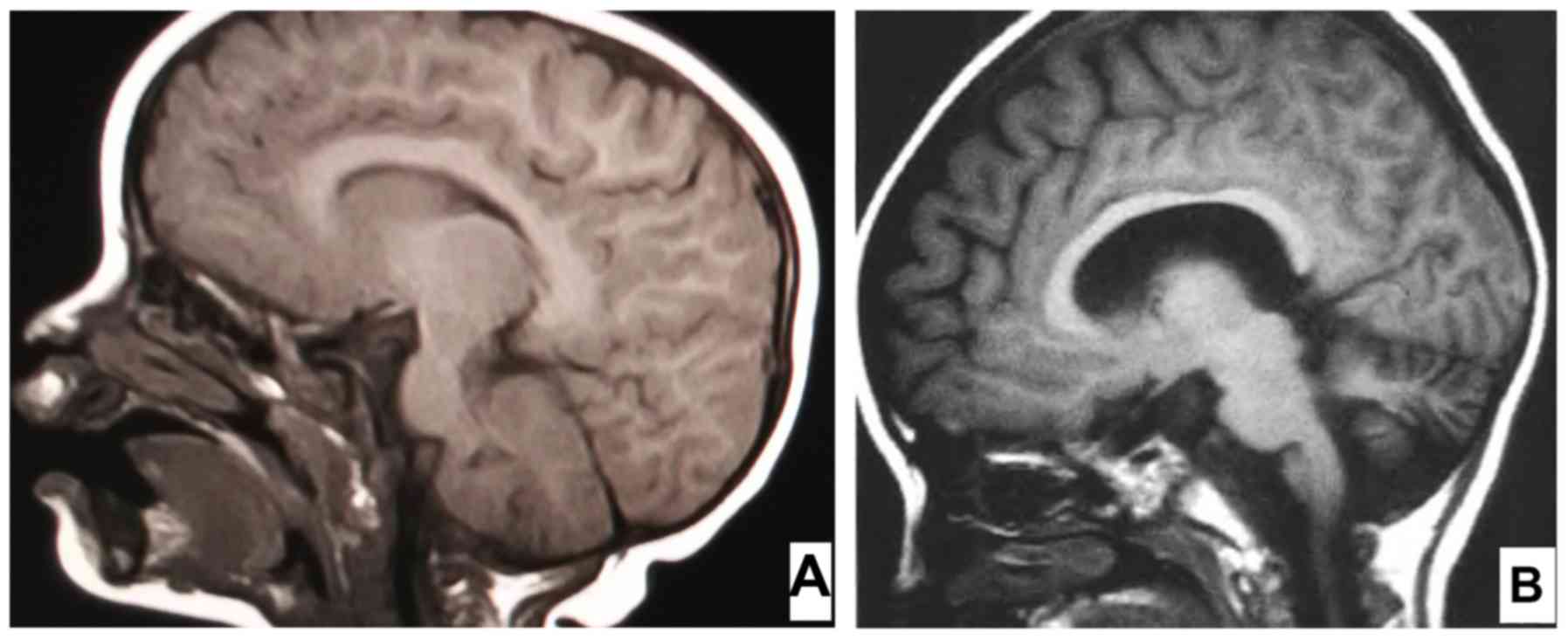
the brain magnetic resonance image (MRI) indicated no cerebellar or
pyramidal tract signs (Fig. 1A).
Electromyography revealed bilateral tibial nerve H reflex latency
and the background of the electroencephalogram (EEG) displayed
high-potential 3–6 Hz waves with no epileptiform discharges. Rapid
global regression of skills with gradually increasing hypotonia was
noted from the age of 18 months, and the auditory brainstem
response (ABR) indicated hearing loss, while brain MRI revealed
cerebellar atrophy with widened folia (Fig. 1B).
Next-generation sequencing (NGS) and
DNA sequence analysis
NGS was performed using an Illumina Hiseq2500
(Illumina, Inc., Santiago, CA, USA). In brief, genomic DNA was
extracted from leucocytes of 2–4 ml peripheral blood using a
BloodGen Midi kit (CWBio, Beijing, China), and was hybridized and
enriched for whole-exome sequencing according to the manufacturer's
protocol. Libraries were generated using an Exomev2.0 kit (Roche
NimbleGen, Inc., Madison, WI, USA) and were measured using the
Illumina Hiseq2500.
Data filtering, mapping and variant
detection
Raw image files were processed using the BclToFastq
(Illumina, Inc.) for base calling and generating the raw data. The
low-quality variations were filtered out by only including those
with a quality score of ≥20 (Q20). The sequencing reads were
aligned to the National Center for Biotechnology Information (NCBI)
human genome 19 (also known as the Genome Reference Consortium
Human Build 37) as a reference by using Burrows Wheeler Aligner
software (5). SAMtools (6) and Pindel (7) were used to analyze single nucleotide
polymorphisms (SNPs) and indels of the sequences, respectively.
Data analysis
Data analysis was performed as follows: i)
Synonymous changes and SNPs with a minor allele frequency of >5%
were removed (http://www.ncbi.nlm.nih.gov/projects/SNP). ii)
Non-synonymous changes were filtered using SIFT software
(http://sift.jcvi.org). iii) The function of the
mutated genes and their association with disease was assessed. iv)
Heredity analysis was performed and included genetic analysis,
clinical symptoms and medical history from the patient and the
patient's parents.
Confirmation of the mutation by Sanger
sequencing
Sanger sequencing was performed to confirm the
mutation of the PLA2G6 gene in the proband. The polymerase chain
reaction (PCR) was performed as previously described (8) and the primers (Sangon Biotech Co.,
Ltd., Shanghai, China) and the length of the PCR products are
displayed in Table I.
 | Table I.Primers of the PLA2G6 gene, polymerase
chain reaction details for 30 cycles and the length of the
respective polymerase chain reaction products. |
Table I.
Primers of the PLA2G6 gene, polymerase
chain reaction details for 30 cycles and the length of the
respective polymerase chain reaction products.
| Name | Sequence (5′-3′) | Denaturation
temperature (°C), duration (sec) | Annealing temperature
(°C), duration (sec) | Extension temperature
(°C), duration (sec) | Product length
(bp) |
|---|
| PLA2G6-1F |
CACTTTAGTTTCCAGGCGTGCTTT | 94, 60 | 55, 30 | 72, 60 | 853 |
| PLA2G6-1R |
GCCGTCCCCAGGTTTATCAAGCAA' |
|
|
|
|
| PLA2G6-2F |
TTTACCTCCCACTCAGTGTTGTTT | 94, 60 | 55, 30 | 72, 30 | 367 |
| PLA2G6-2R |
AACAGAATCAGCTGCCCTTCC |
|
|
|
|
The PCR products were sequenced on an ABI 3730×L
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), analyzed with DNASTAR software (https://www.dnastar.com/) and compared with mRNA
template NM_003560.2 (https://www.ncbi.nlm.nih.gov/nuccore/NM_003560.2)
to verify the base sequence.
Results
NGS
NGS achieved 99.2% coverage of the target sequence
and the average depth was 101.86×. A total of 7,384 mismatch sites
were identified in the sample. The main exome findings for the
patient are presented in Table II.
Mutations of TTC8 (OMIM, 615985), IFT140 (OMIM, 266920) and PSAT1
(OMIM, 616038) were recessive, not leading to disease, and the
observed HTT (OMIM, 143100) mutation was dominant, but clinical
symptoms did not indicate Huntington's disease. It was confirmed
that two mutations of PLA2G6 were pathogenic by clinical symptoms,
mutations and heredity analysis (9).
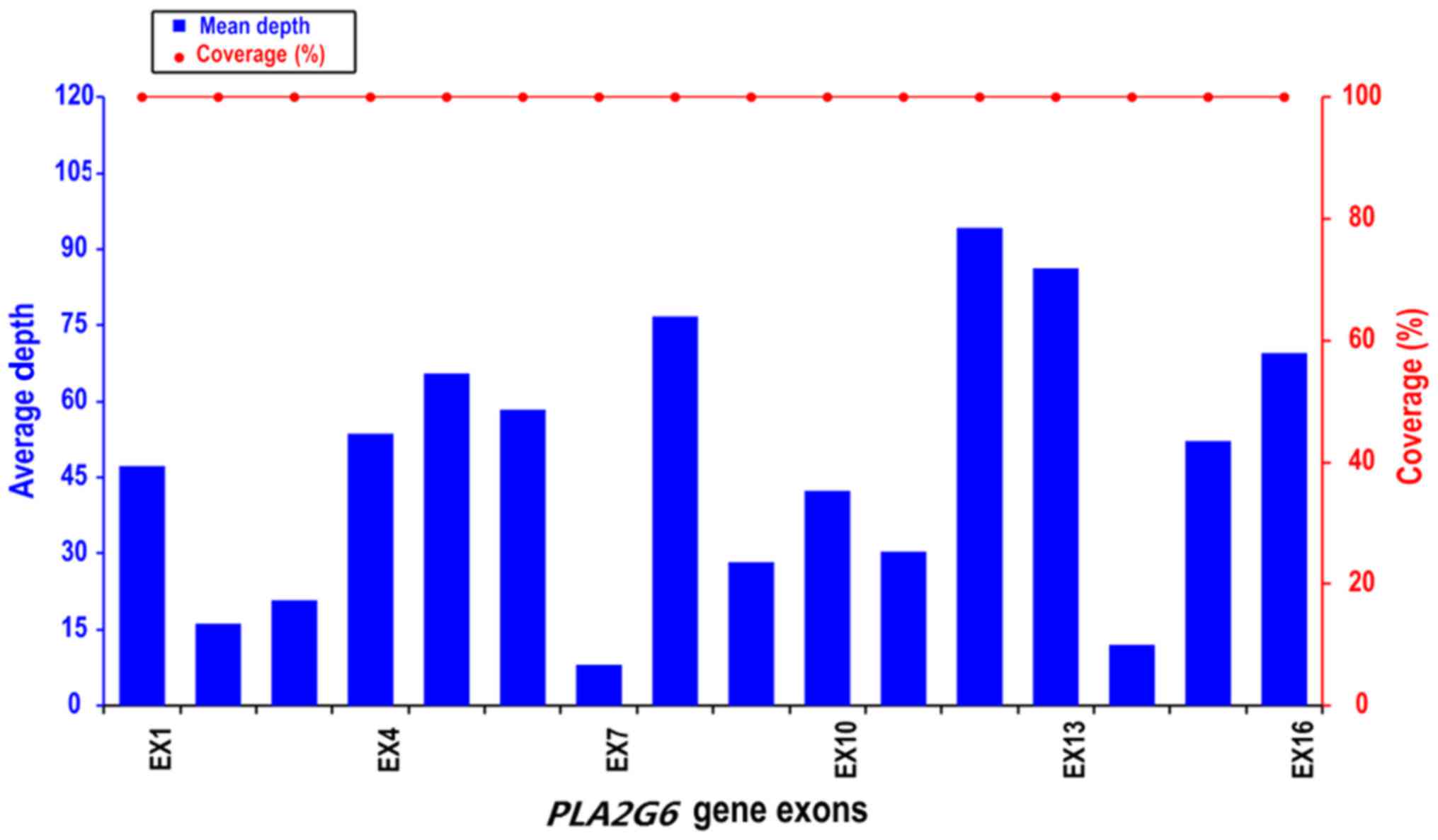
The coverage of PLA2G6 was 100% and the average depth of each exon
of the PLA2G6 gene was >10× (Fig.
2).
| Table II.Major exome analysis results for the
patient. |
Table II.
Major exome analysis results for the
patient.
| Gene | Mutation | Location | Minor allele
frequency | Mode of
inheritance | Associated
syndrome |
|---|
| PLA2G6 | p.M1V |
22:38565433(T>C) | 0 | AR | INAD |
|
| p.E166Gfs32 |
22:38539224-38539225 | 0 | AR | INAD |
| TTC8 | p.I410K | 14:89338678 | 0.001 | AR | Bardet-Biedl
syndrome |
| IFT140 | p.A1330T | 16:1569934 | 0.002 | AR | Mainzer-Saldino
syndrome |
| PSAT1 | p.T156M | 9:80921299 | 0 | AR | Neu-Laxova
syndrome |
| HTT | p.G551E | 4:3129240 | 0.006 | AD | Huntington's
disease |
PLA2G6 gene mutations
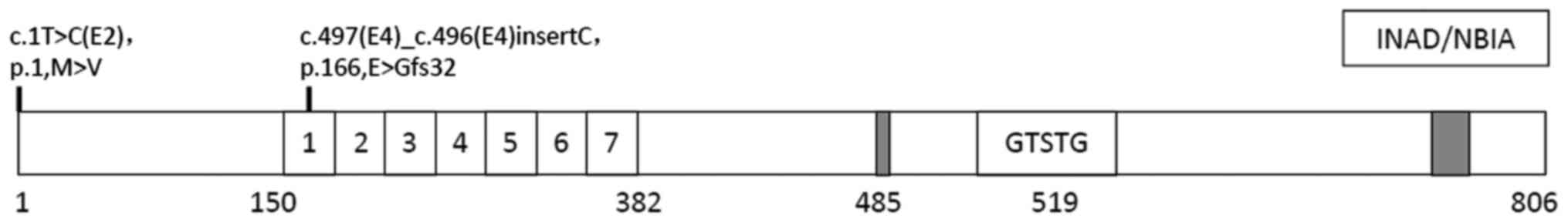
In the proband of the present study, two
heterozygous mutations in PLA2G6 were detected; one was a missense
mutation (p.M1V) in exon 2 and the other was a frame-shift mutation
(p.E166Gfs32) in exon 4. The frame-shift mutation identified in the
proband was only detected in the mother's sample and the missense
mutation was only detected in the father's sample. INAD was
diagnosed according to the clinical symptoms and the results of
genetic testing, and the mutations identified in the present study
were not contained in the SNP (http://www.ncbi.nlm.nih.gov/SNP), HapMap (http://www.hapmap.org), 1000 Genomes Project
(http://www.genomics.cn) and Exome Aggregation
Consortium (ExAC; http://exac.broadinstitute.org/) databases.
Discussion
INAD is a rare progressive neurodegenerative
disorder characterized by pathologic axonal swelling and spheroid
bodies in the CNS, with onset within the first 2 years of life.
Most patients with INAD present with a progressive disorder with
motor and mental deterioration, cerebellar ataxia, marked hypotonia
of the trunk with later bilateral pyramidal tract signs, spastic
tetraplegia, hyperreflexia and early visual disturbances (1).
In the appropriate clinical context, characteristic
brain imaging and fast rhythms on EEG may support the decision to
perform a molecular analysis and avoid skin biopsy to confirm the
diagnosis (9). In previously
published studies, strabismus, optic atrophy and fast rhythms on
EEG are commonly, but not universally reported (10). Prior to the availability of modern
technologies for genetic analysis for the diagnosis if INAD,
neuropathological data were the only resource for the definitive
diagnosis.
In the present study, two mutations of the PLA2G6
gene were identified in the proband. The missense mutation (p.M1V)
in exon 2 replaces the first methionine with valine in the coding
region, while initiation codon mutations affect the initiation
sites of protein-coding genes, and lead to abnormal protein
translation and dysfunction. The frame shift mutation (p.E166Gfs32)
in exon 4 leads to a mutation of the 166th glutamic acid of
glycine, so that coding is terminated after the encoding of 32
amino acids, resulting in a protein short of 662 amino acids, which
lacks the functional domain part with the ankyrin repeat regions
(amino acids 150–382), the GXSXG lipase catalytic site (S519), the
nucleotide binding domain and the calmodulin binding region (amino
acids 747–759) of the protein encoded by PLA2G6, which seriously
affects its function (Fig. 3). Engel
et al (11) reported that
those mutations associated with INAD significantly reduced PLA2G6
phospholipase (iPLA2-VI) activity. PLA2G6 encodes iPLA2-VI, a
calcium-independent phospholipase, which has a crucial role in
maintaining cell membrane homeostasis through phospholipid
remodeling, regulation of apoptosis and catalysis of the hydrolysis
of glycerophospholipids (10,12).
Kinghorn et al (13)
indicated that loss of normal PLA2G6 gene activity leads to lipid
peroxidation, mitochondrial dysfunction and subsequent
mitochondrial membrane abnormalities. Sumi-Akamaru et al
(14) identified a novel
pathological mechanism whereby axons degenerate due to defective
maintenance and rupture of the inner mitochondrial and presynaptic
membranes, which may result in axonal dystrophy.
Zhao et al (15) indicated that the absence of PLA2G6
causes neuroinflammation and Purkinje cell loss, ultimately leading
to cerebellar atrophy. While no obvious abnormities in the
cerebellum were identified in the case of the present study at 14
months of age, the cranial MRI indicated cerebellar atrophy, which
was consistent with the pathology of INAD at 18 months of age. The
results of the two brain MRIs of the proband at 14 and 18 months of
age display obvious differences, indicating a period of rapid
progression of INAD.
Distinct phenotypes associated with mutations in the
same gene may result from the additive influence of genetic and
environmental factors. Alternatively, various single PLA2G6 gene
mutations may primarily determine the phenotype through distinct
effects on protein function, causing either different degrees of
impairment in a single function, or perhaps affecting different
functions of the same protein (11).
The patient's ABR indicated hearing loss; however,
INAD associated with hearing impairments has been rarely reported
in previous studies (16–18). To date, only one other study has
reported on a PLA2G6 mutation leading to INAD with hearing loss
(18). In the above study, two
mutations, namely c.A2047 T at p. K683× and c.T 671 C at p. L224P,
were reported (18). The deafness
may be associated with the mutations of PLA2G6 or have other
causes, which requires further study. According to the 4 studies
available that report on PLA2G6-associated neurodegeneration in
China (19–22), 29 different mutations were identified
in 28 Chinese pediatric patients with PLA2G6-associated
neurodegeneration, of which 14 were novel. However, it remains to
be clarified whether and how PLA2G6 mutations lead to hearing
loss.
INAD is an important pathology/differential
diagnosis to consider in pediatric patients presenting with early
rapid cognitive, motor regression and axial hypotonia. Further
studies are necessary to investigate the correlation between gene
defects and the pathogenesis of INAD. To avoid ultrastructural
studies using peripheral biopsies based on the clinical symptoms,
cranial MRI and screening of mutations in the PLA2G6 gene may be
performed to confirm the diagnosis of INAD (9). The present results on those mutations
indicate that screening for PLA2G6 gene mutations may be utilized
for early diagnosis and genetic counseling, and may be useful to
avoid high-risk pregnancies, as biopsy analysis may confirm
diagnosis.
Acknowledgements
The authors would like to thank Joy Orient from the
Translational Medicine Research Center for the technical
support.
Funding
No funding received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BW performed the experiments. BW, JT and DW analyzed
and interpreted the data and were involved in drafting the
manuscript. JT and DW were involved in critically revising the
manuscript for important intellectual content. All authors read and
approved the manuscript and are accountable for all aspects of the
study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent for the publication of the
patient's data and images was obtained from the guardians.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aicardi J and Castelein P: Infantile
neuroaxonal dystrophy. Brain. 102:727–748. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Larsson PK, Claesson HE and Kennedy BP:
Multiple splice variants of the human calcium-independent
phospholipase A2 and their effect on enzyme activity. J Biol Chem.
273:207–214. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morgan NV, Westaway SK, Morton JE, Gregory
A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et
al: PLA2G6, encoding a phospholipase A2, is mutated in
neurodegenerative disorders with high brain iron. Nat Genet.
38:752–754. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Finster M and Wood M: The Apgar score has
survived the test of time. Anesthesiology. 102:855–857. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye K, Schulz MH, Long Q, Apweiler R and
Ning Z: Pindel: A pattern growth approach to detect break points of
large deletions and medium sized insertions from paired-end short
reads. Bioinformatics. 25:2865–2871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mullis KB, Erlich HA, Arnheim N, Horn GT,
Saiki RK and Scharf SJ: Process for amplifying, detecting,
and/or-cloning nucleic acid sequences. US Patent no. US4683195.
issued. Jul 28–1987.
|
|
9
|
Carrilho I, Santos M, Guimarães A,
Teixeira J, Chorão R, Martins M, Dias C, Gregory A, Westaway S,
Nguyen T, et al: Infantile neuroaxonal dystrophy: What's most
important for the diagnosis? Eur J Paediatr Neurol. 12:491–500.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Illingworth MA, Meyer E, Chong WK, Manzur
AY, Carr LJ, Younis R, Hardy C, McDonald F, Childs AM, Stewart B,
et al: PLA2G6-associated neurodegeneration (PLAN): Further
expansion of the clinical, radiological and mutation spectrum
associated with infantile and atypical childhood-onset disease. Mol
Genet Metab. 112:183–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engel LA, Jing Z, O'brien DE, Sun M and
Kotzbaueret PT: Catalytic function of PLA2G6 is impaired by
mutations associated with infantile neuroaxonal dystrophy but not
dystonia-parkinsonism. PLoS One. 5:e128972010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Z and Turk J: The molecular biology of
the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid
Res Mol Biol. 67:1–33. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kinghorn KJ, Castillo-Quan JI, Bartolome
F, Angelova PR, Li L, Pope S, Cocheme HM, Khan S, Asghari S, Bhatia
KP, et al: Loss of PLA2G6 leads to elevated mitochondrial lipid
peroxidation and mitochondrial dysfunction. Brain. 138:1801–1816.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sumi-Akamaru H, Beck G, Kato S and
Mochizuki H: Neuroaxonal dystrophy in PLA2G6 knockout mice.
Neuropathology. 35:289–302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Z, Wang J, Zhao C, Bi W, Yue Z and Ma
ZA: Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy
characterized by purkinje cell loss and glial cell activation. PLoS
One. 6:e269912011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wisniewski KE, Laure-Kamionowska M, Sher J
and Pitter J: Infantile neuroaxonal dystrophy in an albino girl.
Acta Neuropathol. 66:68–71. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Itoh K, Kawai S, Nishino M, Lee Y, Negishi
H and Itoh H: The clinical and pathological features of siblings
with infantile neuroaxonal dystrophy-early neurological,
radiological, neuroelectrophysiological and neuropathological
characteristics. No To Hattatsu. 24:283–288. 1992.(In Japanese).
PubMed/NCBI
|
|
18
|
Kulkarni SD, Meenal G, Rafat S and Patil
VA: Two unusual cases of PLA2G6-associated neurodegeneration from
India. Ann Indian Acad Neurol. 19:115–118. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Zou Y, Bao X, Wang H, Wang J, Jin H,
Che Y and Tang X: Monozygotic twins with infantile neuroaxonal
dystrophy: A case report and literature review. Exp Ther Med.
12:3387–3389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang P, Gao Z, Jiang Y, Wang J, Zhang F,
Wang S, Yang Y, Xiong H, Zhang Y, Bao X, et al: Follow-up study of
25 Chinese children with PLA2G6-associated neurodegeneration. Eur J
Neurol. 20:322–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y,
Xiong H, Chang X, Bao X, Zhang Y, Xiao J and Wu X: Clinical study
and PLA2G6 mutation screening analysis in Chinese patients with
infantile neuroaxonal dystrophy. Eur J Neurol. 16:240–245. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Wu W, Chen X, Zhang L, Wang X and
Dong G: A novel homozygous mutation in PLA2G6 gene causes infantile
neuroaxonal dystrophy in a case. Chin J Med Genet. 33:64–67.
2016.(In Chinese).
|