Introduction
Hemophagocytic lymphohistiocytosis (HLH), also known
as hemophagocytic syndrome, is a rare, life-threatening
hematological disorder (1). The
occurrence rate of HLH in adults is not well known (1). Despite this, epidemiology data
collected from tertiary medical centers has indicated that the
incidence rate is 1 out of every 2,000 adults (2). However, reported incidence of HLH in
children varies among different studies, which may reflect a
different prevalence within various ethnic groups (3–5). For
example, HLH has been reported to have an incidence of 0.12 per
100,000 children per year in Sweden, with a male to female ratio of
1:1 (4), whereas it was found to be
0.342 per 100,000 in Japan, with a male to female ratio of 0.8:1
(5). HLH is caused by the
uncontrolled proliferation and activation of lymphocytes and
macrophages (1). It is classified as
either familial/primary or acquired/secondary HLH (1). Secondary HLH may be associated with
malignancy, viral infection or autoimmune conditions (1). The current therapeutic strategy
involves the use of immunosuppressive agents; however, current
literature indicates that the mortality rate of patients with
secondary HLH is 50–75% (6).
Atypical rashes may present in certain patients with HLH; however,
to the best of our knowledge annular erythema multiforme-like
eruptions have not previously been reported in cases of HLH.
Herein, the case of a patient who acquired infectious mononucleosis
(IM) and angioimmunoblastic T cell lymphoma (AITL)-associated HLH
at the same time as annular erythema multiforme-like eruptions is
reported. There are three previously reported cases of
AITL-associated HLH (7–9), but these were not accompanied by
annular erythema multiforme-like eruptions. This was a rare case,
and the patient was initially misdiagnosed with erythema multiforme
and drug eruption. The patient was subsequently successfully
diagnosed and treated.
Case report
A 53-year-old male patient, who was diagnosed with
rectal carcinoma and had undergone chemotherapy following
proctectomy for 1 year, suffered from cough and submandibular
lymphadenopathy 9 days prior to admitting to the Dermatology
Department of Beijing Chao-Yang Hospital (Beijing, China) with
swelling and slight pain but no fever. The patient was diagnosed
with acute tonsillitis by otolaryngologists and was initially
treated with oral cefuroxime axetil tablets (dosage not
documented). However, treatment was stopped following taking the
medicine for 4 days due to the condition not entering remission.
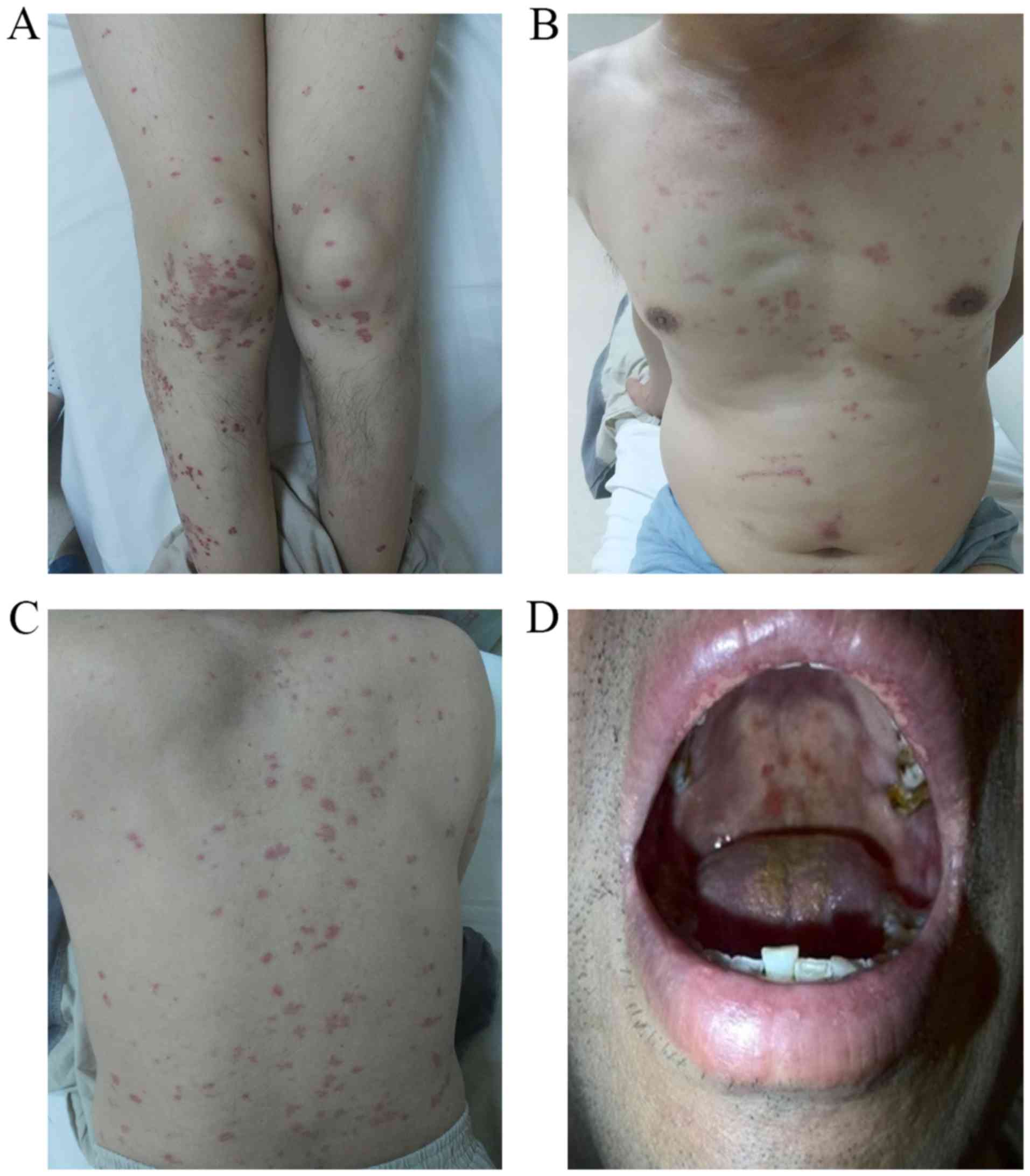
Annular erythema and maculopapular rashes or purpura were observed
accompanied with pruritus on the patient's legs 7 days prior to
admission to the ward (Fig. 1A).
Similar lesions appeared on the patient's chest (Fig. 1B) and back (Fig. 1C) at 4 days prior to admission
without fever. Complete blood cell count was normal, revealing a
white blood cell count of 6.72×109/l (normal,
3.5–9.5×109/l) (10),
monocyte count of 0.48×109/l (normal,
0.1–0.6×109/l) (10),
hemoglobin concentration of 13.7 g/dl (normal, 13.0–17.5 g/dl)
(10) and platelet count of
213.0×109/l (normal, 125.0–350.0×109/l)
(10). Initially, the patient was
diagnosed with erythema multiforme and drug eruption. In primary
care, he was treated with oral prednisolone, antihistamine and
steroid ointment.
Consequently, the rashes did not disappear and fever
developed on the third day with temperature fluctuating between
36.3 and 39.0°C in the ward. Physical examination detected
superficial lymphadenopathy (size, <1×1 cm). Diffuse membranous
tonsillitis appeared and spots of small hemorrhage also appeared on
the hard and soft palates and the connection between the hard and
soft palates (Fig. 1D). Blood cell
count revealed that monocyte content increased when compared with
levels at admission, with a white blood cell count of
8.35×109/l, monocyte count of 1.02×109/l,
hemoglobin concentration of 13.0 g/dl and platelet count of
211.0×109/l. Notably, a population of medium-sized
morphologically atypical lymphocytes in the blood accounted for 9%
of all lymphocytes. The concentrations of liver enzymes were
slightly increased compared with normal ranges; the concentration
of alanine aminotransferase was 52 U/l (normal, 9–50 U/l) (11) and of aspartate aminotransferase was
30 U/l (normal, 15–40 U/l) (11).
The level of triglyceride was normal. Antinuclear antibodies,
anti-double-stranded DNA antibodies and rheumatoid factor were
negative. The level of C-reactive protein was elevated (3.96 mg/dl;
normal, 0–0.8 mg/dl) (12), and the
erythrocyte sedimentation rate was increased (43 mm/h; normal, 2–15
mm/h) (12). Epstein-Barr virus
(EBV) DNA was detected in peripheral blood at a high copy number
(8.42×104/l copies IU/ml; normal,
<5.0×102/l copies IU/ml) by quantitative polymerase
chain reaction (measured in hospital's molecular genetics
department) (13). A serological
examination (measured in hospital's molecular genetics department)
(14) indicated that EBV
immunoglobulin (Ig)G was positive and EBV IgM was negative. Other
serologic profiles, including of EBV viral capsid antigen and
nuclear antigen, were not examined in hospital. The patient refused
skin biopsy, and abdominal ultrasound indicated splenomegaly. As a
result, the patient was diagnosed with IM and was treated with
ganciclovir and prednisone.
After 8 days, the rashes on the patients' limbs and
torso diminished slightly following treatment. However, body
temperature increased repeatedly (maximum, 39.5°C for >7 days)
and sweating was experienced during the night. Furthermore, the
size of supraclavicular lymph nodes increased (>1×1 cm).
Computed tomography scans confirmed hepatosplenomegaly and
lymphadenopathy of multiple nodes in the abdominal, pelvic cavity,
retroperitoneal and the inguinal regions. In addition, blood cell
count as aforementioned, revealed that hemoglobin concentration and
platelet count in the patient continued to decline progressively,
with a hemoglobin concentration of 11.6 g/dl and a minimum platelet
count of 37.0×109/l with 10% atypical lymphocytes. Serum
ferritin and lactate dehydrogenase levels were 785.80 ng/ml and 370
U/l, respectively. The level of EBV DNA increased to
6.0×105/l copies IU/ml. The coagulation profile revealed
prothrombin time of 3.4 sec (normal, 9.6–13.0 sec), activated
partial thromboplastin time of 41.2 sec (normal, 21.0–34.0 sec) and
fibrinogen of 240.8 mg/dl (normal, 170.0–400.0 mg/dl). A
serological examination indicated the presence of polyclonal
gammopathy with a value of 2,280 mg/dl for IgG (normal, 715–1,560
mg/dl), 677 mg/dl for IgM (normal, 82–453 mg/dl) and 1,550 mg/dl
for IgA (normal, 46–304 mg/dl). The bone marrow examination
indicated hemophagocytosis and abnormal lymphocytes. The level of
soluble cluster of differentiation (CD)25 was increased (>44,000
pg/ml; normal <6,400 pg/ml; test result from Beijing Friendship
Hospital, Beijing, China) (15), and
natural killer (NK) cell activity was normal (25.38%; normal
≥15.11%; test result from Beijing Friendship Hospital, Beijing,
China) (16). Therefore, there was a
clear indication of HLH. The patient was subsequently treated with
ganciclovir, dexamethasone and etoposide. At 1 day following
chemotherapy, the platelet count increased to 63×109/l
and the patient exhibited a normal temperature.
Simultaneously, biopsies of the left cervical lymph
node indicated the disappearance of normal structure, infiltration
of eosinophil cells and small to medium-sized lymphoid cells in the
extra membrane and vascular proliferation (Fig. 2A and B). The immunohistochemical
staining of lymphoid infiltrates indicated diffuse positive
staining for CD2, CD3, CD5, CD7 and CD10 as well as weak positive
staining for CD20, paired-box domain 5 and telomerase B (17). The staining for CD21 suggested damage
of follicular dendritic cells and a high expression of Ki-67
(Fig. 2C-L) (17). EBV-encoded RNA in-situ
hybridization (EBER) revealed that the tumor cells were positive
for EBV (18). Therefore, there was
a clear indication of AITL. Consequently, the patient was treated
with etoposide together with cyclophosphamide, doxorubicin,
vincristine and prednisolone (CHOP regimen). The patient was
successfully treated with several courses of chemotherapy, and
clinical manifestations improved. The rashes faded away completely.
The patient was followed up once every 2 weeks for 3 months and he
was deemed to be in good condition.
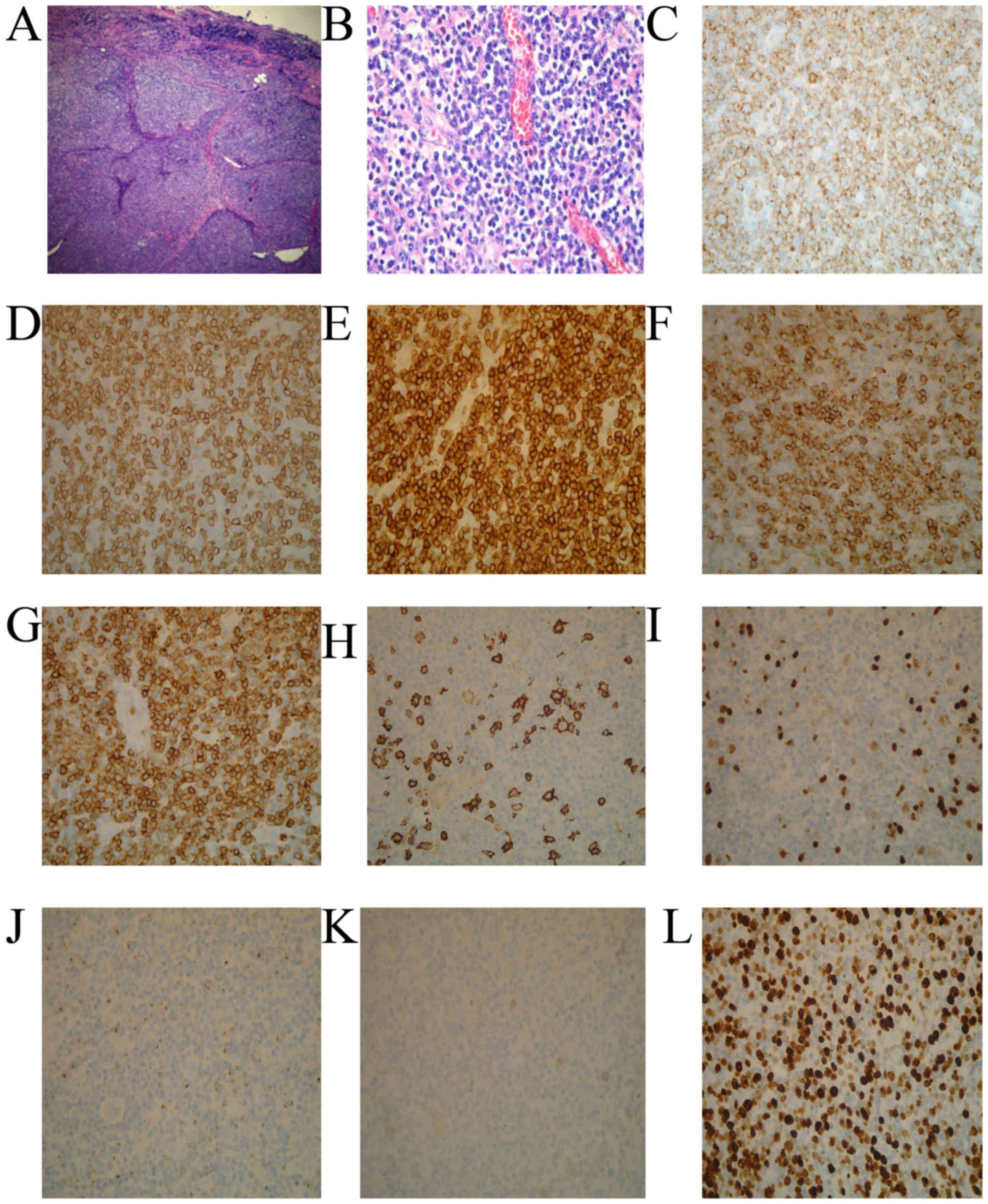 | Figure 2.Histological and immunohistochemical
staining findings of the patient. A biopsy sample taken from the
left cervical lymph node revealed that normal structure had
disappeared, and small to medium-sized atypical lymphoid cells had
infiltrated the lymph node region. (A) H&E staining; original
magnification, ×10. (B) H&E staining; original magnification,
×40. (C-G) On immunohistochemical staining, the lymphoid cells were
diffusely positive for CD2, CD3, CD5, CD7 and CD10 (H&E
staining; original magnification, ×40). (H) CD20, (I) paired-box
domain 5 and (J) telomerase B staining were weak positive (H&E
staining; original magnification, ×40). (K) CD21 staining suggested
follicular dendritic cells net damage (H&E staining; original
magnification, ×40). (L) Proliferative index with Ki-67 stain was
high (H&E staining; original magnification, ×40). H&E,
hematoxylin and eosin; CD, cluster of differentiation. |
Test results, including EBV DNA qPCR, lymph node
biopsies and EBER, were collected from professional clinical
laboratory technicians and professional pathologists from Beijing
Chao-Yang Hospital (Beijing, China). To perform qPCR patient DNA
was extracted. Blood was collected into EDTA-coated tubes and
isolated manually using the QIAamp Blood Mini kit (Qiagen Inc.,
Valencia, CA). Quantitative PCR was then performed using a TaqMan
PCR Core Reagent kit (PerkinElmer, Inc., Waltham, MA, USA). In this
system, a dual-labeled fluorogenic hybridization probe was
included. DNA samples were quantified for EBV DNA using a real-time
qPCR system targeting the BamHI-W fragment region of the EBV
genome (19). The BamHI-W
system utilized the following primers: W-44 forward,
5′-CCCAACACTCCACCACACC-3′ and reverse, W-119
5′-TCTTAGGAGCTGTCCGAGGG-3′. GAPDH was used as the internal
reference gene (forward, 5′-GTCTTCACCACCATGGAGAAGGCT-3′ and
reverse, 5′-CATGCCAGTGAGCTTCCCGTTCA-3′). The dual-labeled
fluorescent probe W-67T [5′-(fluorescent reporter)
CACACACTACACACACCCACCCGTCTC (TAMRA fluorescent dye)-3′;
PerkinElmer, Inc.] (19).
Primer/probe combinations were designed using Primer Express
software (PerkinElmer, Inc.). Fluorescent probes were
custom-synthesized by PerkinElmer, Inc. PCR primers were
synthesized by Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
The thermocycling conditions were as follows: Initial denaturation
for 10 min at 95°C; 40 cycles at 95°C for 15 sec, 56°C for 1 min
and 72°C for 45 sec. Sequence data for the EBV genome were obtained
from the GenBank Sequence Database (https://www.ncbi.nlm.nih.gov/nuccore/V01555) (13). PCR assays were performed in
triplicate. The following quantitative PCR detector systems were
used: STF-Rotor-gene Q (Qiagen, Inc.), 7700 Sequence Detector
(Apple, Inc., Cupertino, CA), and the sequence detection system
software (version 1.7) was developed by PerkinElmer, Inc (13). Pathological biopsies were fixed at
room temperature for 6 h using 10% formalin, dehydrated, embedded
in paraffin, sectioned (thickness, 3–5 µm), de-waxed and stained
with hematoxylin at room temperature for 5–15 min and eosin at room
temperature for 2–3 min as previously described (17). Reagents were from OriGene
Technologies, Inc. (Rockville, MD, USA). Like immunohistochemical
staining, the protocols were as follows: De-waxing, antigen repair
using Histostain™-SP kits (OriGene Technologies, Inc.) and antibody
incubation. The antibodies utilized included: with CD2 (cat. no.
UMAB86), CD3 (cat. no. UM570048), CD5 (cat. no. UM570009), CD7
(cat. no. TA506337), CD10 (cat. no. TA590055), CD20 (cat. no.
UM800065), CD21 (cat. no. TA327627) and Ki-67 (TA801577) all
diluted at 1:100. paired-box domain 5 (cat. no. TA801884) and
telomerase B (cat. no. TA301588) were diluted at 1:50 (all
antibodies were obtained from OriGene Technologies, Inc.) and
hematoxylin staining as previously described (17). EBV-encoded RNA in-situ
hybridization involved sample de-waxing, proteinase K digestion (25
µg/ml, 37°C for 10 min; Merck KGaA, Darmstadt, Germany).
Oligonucleotide probes (5′-CTCCTCCCTAGCAAAACCCTCAGGACGGCG-3′ from
OriGene Technologies, Inc. were labeled using a Labeling kit
(Boehringer Mannheim, S.A., Barcelona, Spain). Labeled probes were
diluted to a concentration of 0.1 µg/ml in hybridisation medium
(50% formamide, 5% dextran sulphate, 2× sodium citrate, sodium
chloride (SSC); all provided by the EBER hybridization kit, OriGene
Technologies, Inc.). Diluted probes were spotted onto tissue
sections and a coverslip was placed on top. Diaminobenzidine
tetrahydrochloride (as obtained from the EBER hybridization kit)
was used as the chromogen (18).
Hybridisation signals were detected using a three layer
ABC-peroxidase technique (Vector Laboratories, Ltd., Peterborough,
UK) (20).
Discussion
HLH is a rare and life-threatening disease, which is
characterized by cytokine storms (1). This hyper-inflammatory reaction can
cause damage to multiple organ systems (1). The causes of mortality in HLH include
multiple organ dysfunction syndrome, massive hemorrhaging and
infectious disease (21).
HLH can be divided into primary (genetic) and
secondary (acquired) HLH. Gene mutations in perforin 1, Unc-13
Homolog D, syntaxin 11 and syntaxin binding protein 2 can result in
damaged cytotoxic function of NK and cytotoxic T cells (1). Primary HLH often occurs in children
(22). However, it has been
demonstrated to also occur in adolescents and adults (22). Infection is a common cause of
secondary HLH, particularly EBV infections (1). Of note, hormones and antiviral
treatments are often effective against acute EBV infections
(1). Secondary HLH, which is
triggered by tumors, typically occurs in adults (~45% of cases)
(23). AITL, a type of T-cell
non-Hodgkin's lymphoma, is frequently accompanied by progressive
systemic symptoms, including high fever, cytopenia, body weight
loss and night sweats (24,25). AITL, unlike other T cell lymphomas,
can be associated with a profound immune deficiency that is caused
by chemotherapy or immunotherapy, which leads to the activation of
EBV (26). In particular, EBV
infections have been reported predominantly in patients with AITL
(27). Zhou et al (28) previously reported that EBV
infections, as a consequence of AITL, may be reactivated in the
presence of immunodeficiency rather than contributing to the
pathogenesis of the disease itself (28). AITL-associated HLH has a poor
prognosis due to continuous disease aggravation, which leads to an
increased risk for opportunistic infections compared with
EBV-associated HLH (29). The
present case demonstrated that AITL was the cause for triggering
HLH. Due to immune deficiency that was induced by rectal carcinoma
chemotherapy, the EBV infection in the patient was activated. There
are three previously reported cases of AITL-associated HLH
(Table I) (7–9).
 | Table I.List of reported patients with
angioimmunoblastic T cell lymphoma associated with Hemophagocytic
lymphohistiocytosis, clinical manifestation, treatment, and
outcomes. |
Table I.
List of reported patients with
angioimmunoblastic T cell lymphoma associated with Hemophagocytic
lymphohistiocytosis, clinical manifestation, treatment, and
outcomes.
| Age, years/sex | Clinical
manifestation | Therapy | Outcome | Reference |
|---|
| 58/F | Fever, pharyngeal
pain, lymphadenopathy, splenomegaly | CHOP, fludarabine,
cyclosporine A, allogeneic peripheral blood stem cell
transplantation | Successfully
treated | 7 |
| 57/M | Fever, neutropenia,
thrombocytopenia, lymphadenopathy | Cyclophosphamide,
prednisolone, etoposide, cicloporin | Succumbed to
multiorgan failure | 8 |
| 62/F | Fever,
lymphadenopathy, hepatosplenomegaly, loss of weight | CHOP, mesna,
ifosfamide, mitoxantrone, etoposide; allogeneic hematopoietic stem
cell transplantation | Successfully
treated | 9 |
Skin manifestations occur in 24–40% cases of genetic
HLH and 6–65% of cases in acquired HLH (28). Reported cutaneous findings have
included erythematous rashes, macules, edema, panniculitis,
morbilliform erythema, petechiae and purpura (30,31). The
lesions in HLH are not characteristic; the most common cutaneous
manifestations are panniculitis and purpura (32,33).
Cutaneous manifestations that are associated with HLH are
classified into three types. The manifestation may be specific to
the underlying malignancy (cutaneous lymphoma or systemic disease),
reflect the biological consequences of HLH (thrombopenic purpura or
conjunctival jaundice) or can be a generalized, transient,
nonpruritic, maculopapular rash (34). However, a case with AITL-associated
HLH and annular erythema multiforme-like eruptions has not been
previously reported, to the best of our knowledge.
The occurrence of AITL-associated HLH with annular
erythema multiforme-like eruptions in a patient is highly rare
worldwide, which led to the misdiagnosis of erythema multiforme.
From this patient and the other 3 reported patients with
AITL-associated HLH, it was observed that AITL-associated HLH
occurred in people with an age of >50 years. CHOP and etoposide
combined with allogeneic hematopoietic stem cell transplantation
may be effective in the early stages of the disease.
In conclusion, this present case of AITL-associated
HLH with annular erythema multiforme-like rashes provides novel
understanding for clinical diagnosis of the disease. Without rapid
diagnosis and early treatment, AITL-associated HLH may lead to
short survival times.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science and Technology Key Projects (grant no. 2017ZX09304029004),
China.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, CT and YH designed the present study and drafted
the manuscript. YT, SP and LZ collected the clinical and imaging
data. TW analyzed and interpreted the patient data regarding the
hematological disease. CT and LZ were major contributors in writing
and revising the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The patient provided informed written consent for
their participation.
Patient consent for publication
The patient provided informed written consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Samkari H and Berliner N:
Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 13:27–49.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parikh SA, Kapoor P, Letendre L, Kumar S
and Wolanskyj AP: Prognostic factors and outcomes of adults with
hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 89:484–492.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gholam C, Grigoriadou S, Gilmour KC and
Gaspar HB: Familial haemophagocytic lymphohistiocytosis: Advances
in the genetic basis, diagnosis and management. Clin Exp Immunol.
163:271–283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Henter JI, Elinder G, Soder O and Ost A:
Incidence in Sweden and clinical features of familial
hemophagocytic lymphohistiocytosis. Acta Paediatr Scand.
80:428–435. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishii E, Ohga S, Tanimura M, Imashuku S,
Sako M, Mizutani S and Miyazaki S: Clinical and epidemiologic
studies of familial hemophagocytic lymphohistiocytosis in Japan.
Med Pediatr Oncol. 30:276–283. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schram AM and Berliner N: How I treat
hemophagocytic lymphohistiocytosis in the adult patient. Blood.
125:2908–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsumura Y, Kuroda J, Shimura Y, Kiyota
M, Yamamoto-Sugitani M, Kobayashi T, Matsumoto Y, Horiike S and
Taniwaki M: Cyclosporine a and reduced-intensity conditioning
allogeneic stem cell transplantation for relapsed
angioimmunoblastic t cell lymphoma with hemophagocytic syndrome.
Intern Med. 51:2785–2787. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vella JE and El-Daly H: Hemophagocytic
lymphohistiocytosis in a patient with angioimmunoblastic lymphoma:
A case report and review of the literature. Int J Surg Pathol.
20:606–609. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu JT, Hwang WL, Wang RC and Teng CL:
Reduced intensity conditioning allogeneic hematopoietic stem cell
transplant could be beneficial to angioimmunoblastic T-cell
lymphoma patients with hemophagocytic lymphohistiocytosis. Ann
Hematol. 91:805–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu X, Zhao M, Pan B, Zhang J, Peng M, Wang
L, Hao X, Huang X, Mu R, Guo W, et al: Complete blood count
reference intervals for healthy Han Chinese adults. PLoS One.
10:e01196692015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shinya S, Masaru A, Akira H, Eisaku H and
Susumu O: Development of an assay of seven biochemical items,
HbA1c, and hematocrit using a small amount of blood collected from
the fingertip. Clin Chim Acta. 413:192–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Crowson CS, Rahman MU and Matteson EL:
Which measure of inflammation to use? A comparison of erythrocyte
sedimentation rate and C-reactive protein measurements from
randomized clinical trials of golimumab in rheumatoid arthritis. J
Rheumatol. 36:1606–1610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lo YM, Chan LY, Lo KW, Leung SF, Zhang J,
Chan AT, Lee JC, Hjelm NM, Johnson PJ and Huang DP: Quantitative
analysis of cell-free Epstein-Barr virus DNA in plasma of patients
with nasopharyngeal carcinoma. Cancer Res. 59:1188–1191.
1999.PubMed/NCBI
|
|
14
|
Gu AD, Mo HY, Xie YB, Peng RJ, Bei JX,
Peng J, Li MY, Chen LZ, Feng QS, Jia WH and Zeng YX: Evaluation of
a multianalyte profiling assay and an enzyme-linked immunosorbent
assay for serological examination of Epstein-Barr virus-specific
antibody responses in diagnosis of nasopharyngeal carcinoma. Clin
Vaccine Immunol. 15:1684–1688. 2005. View Article : Google Scholar
|
|
15
|
Oboshi W, Aki K, Tada T, Watanabe T,
Yukimasa N, Ueno I, Saito K and Hosoi E: Flow cytometric evaluation
of surface CD56 expression on activated natural killer cells as
functional marker. J Med Invest. 63:199–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Valiathan R, Lewis JE, Melillo AB, Leonard
S, Ali KH and Asthana D: Evaluation of a flow cytometry-based assay
for natural killer cell activity in clinical settings. Scand J
Immunol. 75:455–462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zu Y, Steinberg SM, Campo E, Hans CP,
Weisenburger DD, Braziel RM, Delabie J, Gascoyne RD,
Muller-Hermlink K, Pittaluga S, et al: Validation of tissue
microarray immunohistochemistry staining and interpretation in
diffuse large B-cell lymphoma. Leuk Lymphoma. 46:693–701. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Liu X and Chen Y: Adult systemic
Epstein-Barr virus-positive T-cell lymphoproliferative disease: A
case report. Exp Ther Med. 10:1025–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baer R, Bankier AT, Biggin MD, Deininger
PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC,
Séguin C, et al: DNA sequence and expression of the B95-8
Epstein-Barr virus genome. Nature. 310:207–211. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khan G, Coates PJ, Kangro HO and Slavin G:
Epstein Barr virus (EBV) encoded small RNAs: Targets for detection
by in situ hybridisation with oligonucleotide probes. J Clin
Pathol. 45:616–620. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smith KJ, Skelton HG III, Giblin WL and
James WD: Cutaneous lesions of hemophagocytic syndrome in a patient
with T-cell lymphoma and active Epstein-Barr infection. J Am Acad
Dermatol. 25:919–924. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jordan MB, Allen CE, Weitzman S,
Filipovich AH and McClain KL: How I treat hemophagocytic
lymphohistiocytosis. Blood. 118:4041–4052. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allen CE and McClain KL: Pathophysiology
and epidemiology of hemophagocytic lymphohistiocytosis. Hematology
Am Soc Hematol Educ Program. 2015:177–182. 2015.PubMed/NCBI
|
|
24
|
Takahashi N, Chubachi A, Kume M, Hatano Y,
Komatsuda A, Kawabata Y, Yanagiya N, Ichikawa Y, Miura AB and Miura
I: A clinical analysis of 52 adult patients with hemophagocytic
syndrome: The prognostic significance of the underlying diseases.
Int J Hematol. 74:209–213. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koh MJ, Sadarangani SP, Chan YC, Chan MY,
Tan AM, Tan SH, Tay YK and Ng SB: Aggressive subcutaneous
panniculitis-like T-cell lymphoma with hemophagocytosis in two
children (subcutaneous panniculitis-like T-cell lymphoma). J Am
Acad Dermatol. 61:875–881. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Castillo JJ, Beltran BE, Bibas M, Bower M,
Collins JA, Cwynarski K, Diez-Martin JL, Hernandez-Ilizaliturri F,
Horwitz SM, Montoto S, et al: Prognostic factors in patients with
HIV-associated peripheral T-cell lymphoma: A multicenter study. Am
J Hematol. 86:256–261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zettl A, Lee SS, Rüdiger T, Starostik P,
Marino M, Kirchner T, Ott M, Müller-Hermelink HK and Ott G:
Epstein-Barr virus-associated B-Cell lymphoproliferative disorders
in angioimmunoblastic T-cell lymphoma and peripheral T cell
lymphoma, unspecified. Am J Clin Pathol. 117:368–379. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Y, Attygalle AD, Chuang SS, Diss T,
Ye H, Liu H, Hamoudi RA, Munson P, Bacon CM, Dogan A and Du MQ:
Angioimmunoblastic T-cell lymphoma: Histological progression
associates with EBV and HHV6B viral load. Br J Haematol. 138:44–53.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brown HA, Macon WR, Kurtin PJ and Gibson
LE: Cutaneous involvement by angioimmunoblastic T-cell lymphoma
with remarkable heterogeneous Epstein-Barr virus expression. J
Cutan Pathol. 28:432–438. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zerah ML and DeWitt CA: Cutaneous findings
in hemophagocytic lymphohistiocytosis. Dermatology. 230:234–243.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ariffin H, Lum SH, Cheok SA, Shekhar K,
Ariffin WA, Chan LL and Lin HP: Haemophagocytic lymphohistiocytosis
in Malaysian children. J Paediatr Child Health. 41:136–139. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith KJ, Skelton HG, Yeager J, Angritt P,
Wagner K, James WD, Giblin WJ and Lupton GP: Cutaneous
histopathologic, immunohistochemical, and clinical manifestations
inpatients with hemophagocytic syndrome. Military medical
consortium for applied retroviral research (MMCARR). Arch Dermatol.
128:193–200. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakai H, Otsubo S, Miura T and Iizuka H:
Hemophagocytic syndrome presenting with a facial erythema in a
patient with systemic lupus erythematosus. J Am Acad Dermatol.
57:S111–S114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fardet L, Galicier L, Vignon-Pennamen MD,
Regnier S, Noguera ME, de Labarthe A, Raffoux E, Martinez V, Buyse
S, Viguier M, et al: Frequency, clinicalfeatures and prognosis of
cutaneous manifestations in adult patients with reactive
haemophagocytic syndrome. Br J Dermatol. 162:547–553. 2010.
View Article : Google Scholar : PubMed/NCBI
|