Introduction
Treacher Collins syndrome (TCS) is a severe
congenital disorder characterized by craniofacial malformation that
occurs in 1 out of 50,000 live births (1–4). The
condition was first described by Thompson in 1846, however TCS was
named after E. Treacher Collins, who described the essential
components of the condition in 1900 (5). The first extensive review of the
condition was performed by Franceschetti and Klein in 1949, who
used the term mandibulofacial dysostosis (MFD) to describe relative
clinical features (6–8). The major characteristics of TCS include
cleft palate, hypoplasia of the facial bones, the mandible and
zygomatic complex, downward slanting of the palpebral fissures and
malformation of the external and middle ear (9–12).
Patients affected by TCS require management strategies or treatment
plans for hearing, respiration, a variety of malformations, bad
overall quality of life and mental health, which describes a great
burden to individuals, families and society (13).
TCOF1 mutations occur in >90% of cases of
TCS and are inherited via an autosomal dominant pattern (12). Over the past decade, two additional
gene mutations have been reported in <2% of TCS patients:
POLR1D, which is autosomal dominant, and POLR1C,
which is inherited via autosomal recessive pattern (14–17).
Furthermore, >60% of patients with TCS have no previous family
history of the disease and TCS arises as the result of de
novo mutations (6,17). Affected individuals may transmit the
defect to each child with a 50% probability according to Mendelian
laws of genetics, which emphasizes the importance of genetic
counseling to affected individuals (5). In the present study, the clinical
findings and molecular diagnosis of a Chinese family with TCS are
reported.
Materials and methods
Patients
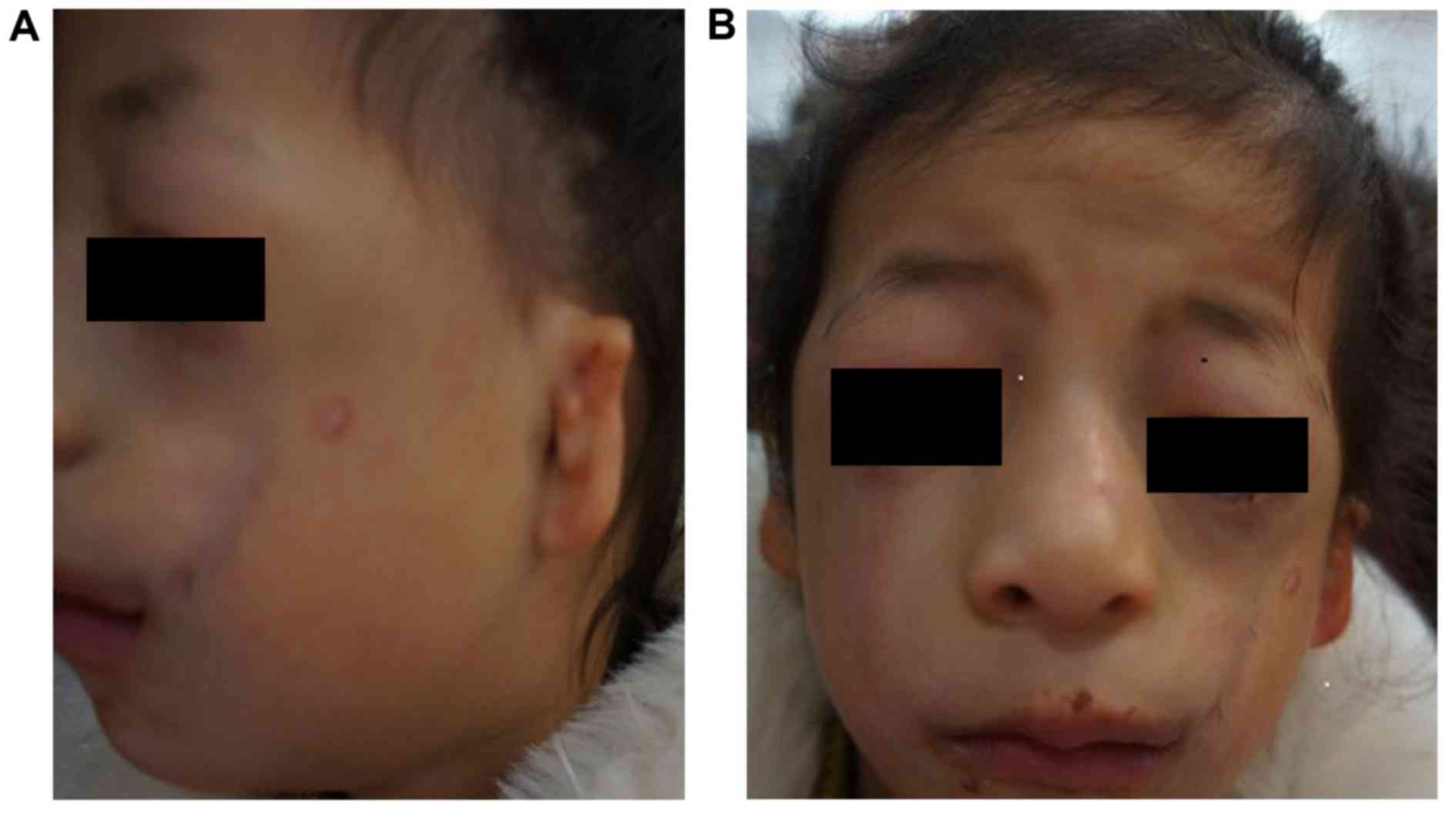
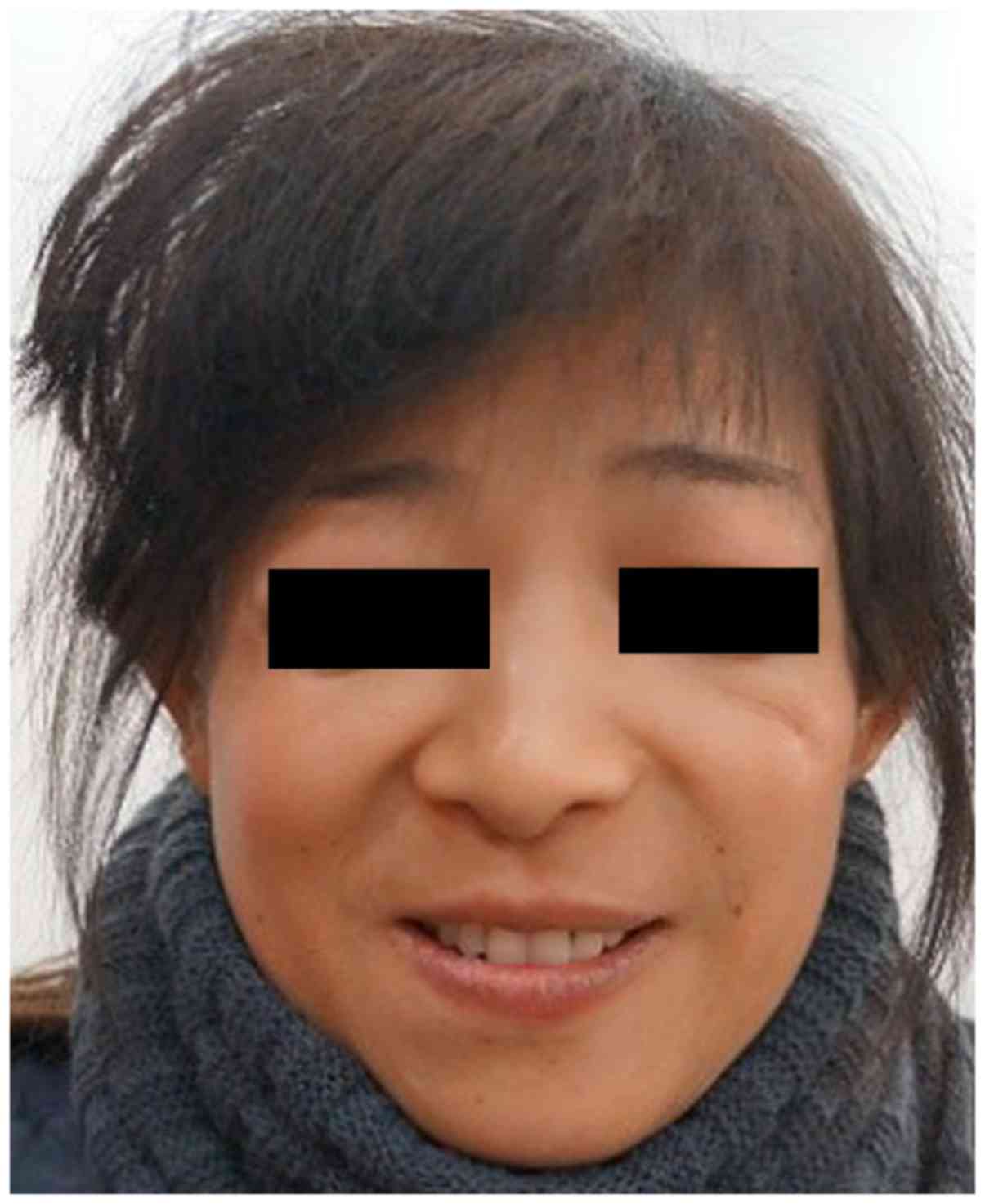
A 9-year-old Chinese girl (the proband; Fig. 1) and her 40-year-old mother (Fig. 2) from Jiangsu, China, were diagnosed
with TCS based on a physical examination in December 2013 at the
Department of Otolaryngology Head and Neck Surgery, Chinese PLA
97th Hospital (Beijing, China). No craniofacial abnormalities were
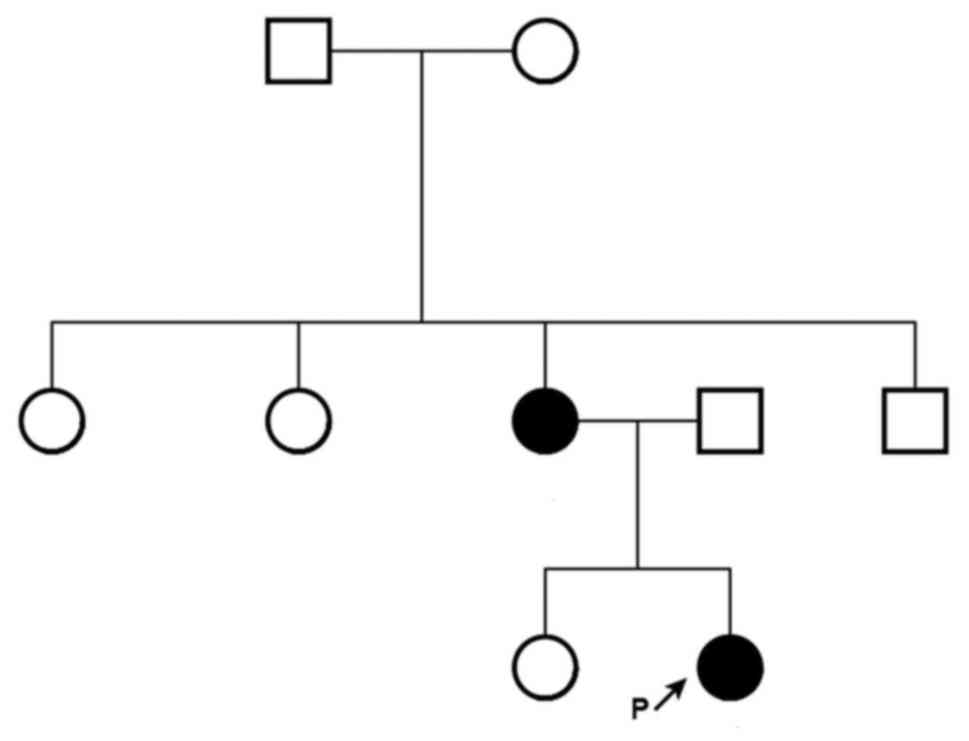
apparent in the proband's father, sister or maternal grandfather,
grandmother, aunts or uncle.
Based on the physical examination of the proband,
the following clinical features were observed: Conductive hearing
loss, microtia, hypoplasia of the middle ear with concomitant
atresia of the external auditory canal, narrow nasal cavity, soft
palate drooping downward, downward slanting of the eyelids,
hypoplasia of the zygomatic bone, mandibular hypoplasia, coloboma
with eyelashes absent in the medial part of the eyelids and funnel
chest. Similar features, but with a lower level of clinical
severity, assessed by slight deformity, including mandibular and
zygomatic hypoplasia; partial absence of lower eyelashes; and
coloboma of the lower lateral eyelid, were detected in her mother.
In addition, the proband and her mother used hearing aids and were
undergoing speech therapy. The family pedigree is presented in
Fig. 3.
The present study was approved by the General
Hospital of PLA Clinical Human Research Ethics Committee (Beijing,
China). All participants provided written informed consent and all
procedures complied with the Declaration of Helsinki.
Sanger sequencing
Following the physical examination, peripheral blood
samples (3–5 ml) were collected from the elbow vein of the
subjects, including the proband and the sister, father, mother,
maternal grandfather and maternal grandmother. Blood samples were
stored at 4°C for DNA extraction within 3 days or at −80°C for a
longer storage times. DNA was extracted from peripheral blood
samples using the AxyPrep DNA blood Midi kit (Axygen; Corning
Incorporated, Corning, NY, USA) according to the manufacturer's
protocol. All TCOF1 exons were sequenced to identify the
causative mutation in the proband. A total of 27 exons of
TCOF1 were amplified using polymerase chain reaction (PCR)
under optimal conditions. Specific primers were designed using
Primer3-v.0.4.0 online software (http://bioinfo.ut.ee/) (Table I).
 | Table I.Self-designed primers used for
TCOF1 gene amplification and sequencing. |
Table I.
Self-designed primers used for
TCOF1 gene amplification and sequencing.
| Exon | Direction | Sequence
(5′-3′) | Length (bp) |
|---|
| 1 | Forward |
GAAAGAGGAGCCGGAAGTGT | 397 |
|
| Reverse |
ACTGAAGTCGCAGTGGGAAG |
|
| 2 | Forward |
CCTCTTCTGAACCACCTGTCTA | 233 |
|
| Reverse |
CTTATTCCAAGCTGAGTTGCTT |
|
| 3 | Forward |
TGCCTATACTGTGTTTTCACCA | 392 |
|
| Reverse |
CCCAGGGTCTTTTAGGTCTTCT |
|
| 4 | Forward |
ACAGAGCTCATTCCTGCAAGTC | 381 |
|
| Reverse |
TAAGATCCCACAACTGGTGACA |
|
| 5 | Forward |
TTGAAGGGGTAGTTTACCCAAA | 275 |
|
| Reverse |
CCCTCGTCTAGGTGATGAGAAA |
|
| 6 | Forward |
CTTTGATGAGCAGCTGGTTT | 228 |
|
| Reverse |
AGGTTCCTGGAAGGGTTAGAG |
|
| 6A | Forward |
AAGCCTTGTGTACTTTTCTGGA | 486 |
|
| Reverse |
AGAGGTGCTCATGGCAGAGT |
|
| 7 | Forward |
GTGTGGCCAAAGTATCAGTCAA | 497 |
|
| Reverse |
ACACAGTGAGAGGGGAGTAAGG |
|
| 8 | Forward |
GGACTTGTTCTCCCACTCTGG | 500 |
|
| Reverse |
GAAACAGGATGAGGGGAGAG |
|
| 9 | Forward |
GAATCGGACAGTGAGGAGGAG | 455 |
|
| Reverse |
GGAAAAGTCAAAACCACAGGAG |
|
| 10 | Forward |
CCTGTGGTTTTGACTTTTCCTC | 500 |
|
| Reverse |
GAGATACACAGGATCGGGAGAG |
|
| 11 | Forward |
ACCTCACACTGGGACTCTGTCT | 480 |
|
| Reverse |
GGAATTTTCAGAGCTGGTTTTG |
|
| 12 | Forward |
ATGGACAACTCGGAGAGCAG | 587 |
|
| Reverse |
GACAAGGGGAAGAGAGGTGTC |
|
| 13 | Forward |
GTGAGGCCTGTGTTTTCTGG | 400 |
|
| Reverse |
CTGAGGCTTCTGCACACCTG |
|
| 14 | Forward |
CTCAGGTTCACACGCCTATTG | 399 |
|
| Reverse |
CCCCACTATGGCACAACTCT |
|
| 15 | Forward |
GGTAGAGAGGAGGACCAGTCAC | 485 |
|
| Reverse |
AGCTCTGATCTGGTGGGTCTT |
|
| 16 | Forward |
TAACACCTTTGCCACATCCA | 388 |
|
| Reverse |
GCCTCCCAAAGTGCTAGGAT |
|
| 16A | Forward |
CCGACCACGTGCTTATCC | 246 |
|
| Reverse |
ATGGCGAGATTTTCCCTATG |
|
| 17 | Forward |
GTGGACCCTTTGCCTTGTAA | 373 |
|
| Reverse |
ACTCAGCCAGTGTCCTGTCC |
|
| 18 | Forward |
GCTCTAGATCACCAGCACAGG | 393 |
|
| Reverse |
TAGGAGGCCAGAAAGCCTCT |
|
| 19 | Forward |
CAGTTTTGCCCCTTTGACTG | 359 |
|
| Reverse |
CAAACCAAGTGCAGAGGTCA |
|
| 20 | Forward |
CATGTGTGCCCCATCTAACA | 474 |
|
| Reverse |
TACAGGTGGGGAAACTGAGG |
|
| 21 | Forward |
GTGAGGGACCTGCAGAGAGA | 270 |
|
| Reverse |
CTGAGGGATCGGGTAGACAG |
|
| 22 | Forward |
AGGGCAGGGTGATCCTAGAG | 298 |
|
| Reverse |
CTGTTTTAGGGGACAACATGC |
|
| 23 | Forward |
ATTGGTGGAAAGGTGTGAGC | 837 |
|
| Reverse |
AGGAATGAGACCAGGTGCTG |
|
| 24 | Forward |
CTGGGATTGCAGGAATGAAC | 398 |
|
| Reverse |
GGTGTGTCACAACCCCTGAC |
|
| 25 | Forward |
CGCTGCAGACCCAGTATCTA | 250 |
|
| Reverse |
TCAGGTCTGCCTGGCTCTCT |
|
PCR amplification was performed in a 25 µl reaction
volume using the Taq PCR Master mix (Biomed Gene Technology Co.,
Ltd., Beijing, China) according to the manufacturer's protocol.
Thermocycling conditions were as follows: Denaturation at 95°C for
5 min followed by 14 cycles of 95°C for 30 sec, annealing at 60°C
for 30 sec and extension at 72°C for 45 sec, followed by 21 cycles
at 95°C for 30 sec, annealing at 56°C for 30 sec and extension at
72°C for 45 sec. The reaction was completed with a final extension
step at 72°C for 7 min. PCR products were purified and sequenced.
Sequencing was performed according to the manufacture's protocol
using an ABI PRISM Big Dye Terminator v3.1 Cycle Sequencing-ready
Reaction kit (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with an ABI 3130 Genetic analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
Mutational analysis
Sanger sequencing reads were mapped to a human
genome reference sequence (hg19; University of California, Santa
Cruz, CA, USA) using the Mutation Surveyor (v.5.0.2; SoftGenetics,
LLC, State College, PA, USA). Consequently, the frequency of each
variant was obtained from dbSNP database (version 132, http://www.ncbi.nlm.nih.gov/snp/); all variants
with a frequency >1% were filtered. Finally, the possible
causative variants were predicted using the combined annotation
dependent depletion tool (http://cadd.gs.washington.edu/) for missense variants.
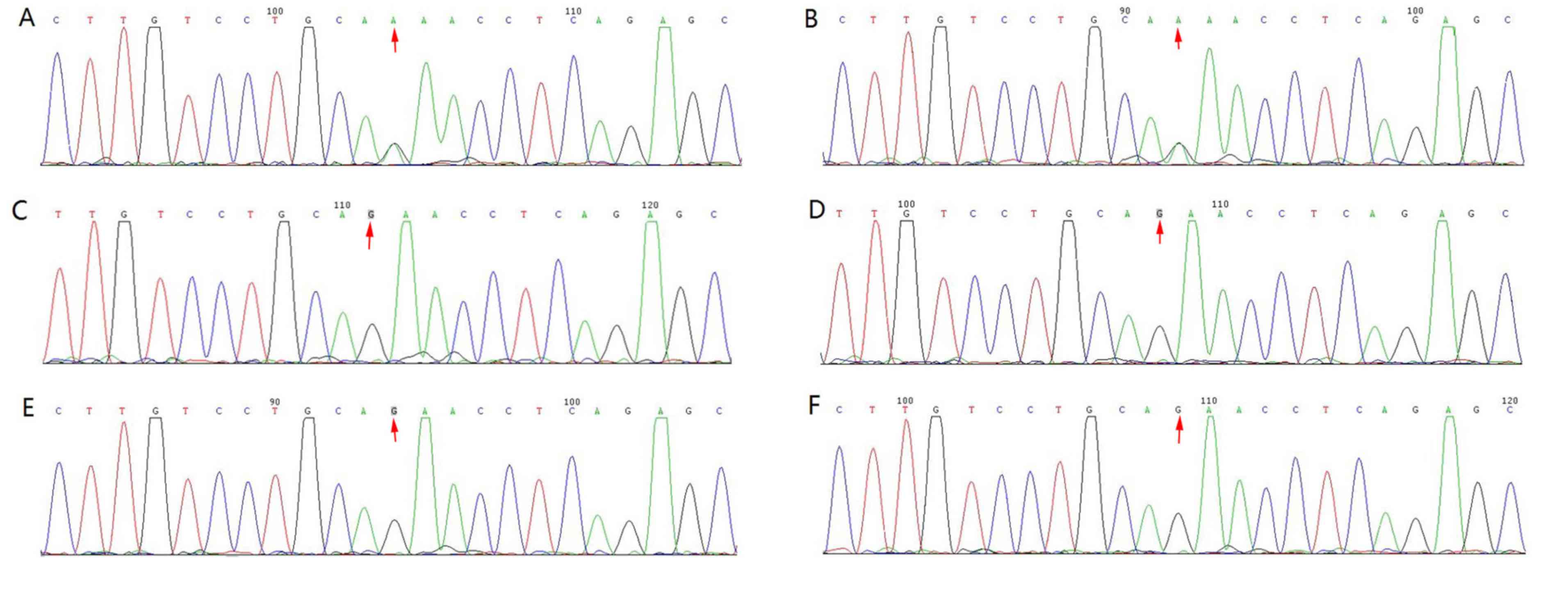
To validate candidate mutations, phenotype-genotype co-segregation
analysis was performed in family members, including the proband and
the sister, father, mother, maternal grandfather and maternal
grandmother, using the Sanger sequencing results (Figs. 3 and 4).
Results
Clinical features
Prominent TCS features were observed in the proband
(Fig. 1). Furthermore, similar
features with lower clinical severity were observed in her mother,
including conductive hearing loss and deformities in face and ear
(Fig. 2).
Sequencing and diagnosis
One novel heterozygous mutation was identified in
the TCOF1 exon 3 splicing site c.165-1G>A (Fig. 4) of the proband and her mother.
Furthermore, segregation analysis confirmed the co-segregation of
this TCOF1 exon 3 coding region mutation (Fig. 4). No apparent craniofacial
abnormalities were observed in the other family members assessed.
In addition, their hearing was normal and there were no changes in
this coding region post-sequencing (Fig.
4).
Discussion
TCS is associated with autosomal dominant mutations
in the TCOF1 gene, which is located on chromosome 5q32-q33
(6,10,18).
TCOF1 encodes a 144 kDa nucleolar phosphoprotein, Treacle,
which serves a role in ribosomal DNA gene transcription via
interacting with upstream binding factors and RNA polymerase I in
the nucleolus (2). TCOF1 is
broadly expressed throughout the embryo, with particularly strong
activity in the neuroepithelium where it serves an essential role
in cell survival (2,11,19).
Treacle participates in ribosome biogenesis by controlling
preribosomal (pre-r)RNA synthesis or by pre-rRNA processing
(8,12). Dixon et al (11) suggested that general cranio skeletal
hypoplasia observed in individuals with TCS is caused by a
deficiency in neural crest cells, rather than neural crest cell
migration defect. TCOF1/treacle is essential for neural
crest cell formation, neuroepithelial survival and neural crest
cell proliferation (11,20). However, the biological role of
Treacle remains to be fully elucidated.
It was previously believed that the TCOF1
gene included 25 exons, 49–561 bp in length (21). In 2004, two additional exons were
discovered by So et al (22):
6A, 231 bp, situated between exons 6 and 7; and 16A, 108 bp,
localized between exons 16 and 17 (21). Pathogenic mutations in the
TCOF1 gene are spread throughout its coding region and
typically comprise point mutations or small frame shift deletions
and insertions, the majority of which are family-specific (23). The existence of mutational hot spots
in TCOF1 has been suggested, indicating that exons 23 and 24
are responsible for ~1/3 of all known pathogenic changes (23). The majority of mutations responsible
for TCS are localized in exons, mainly in the hot spots of exons
10, 15, 16, 23 and 24 (21,24). The TCOF1 gene mutations
include missense, nonsense, small deletions and duplications. The
most common classes of TCOF1 alleles are small deletions
(60%) and duplications (25%), resulting in frame shift (17).
Multiple exons have been identified within the
TCOF1 gene and different splicing patterns result in several
variants of the mutant gene. So far, >200 mutations have been
identified (2). Combined analysis of
the variants and clinical features has not revealed a clear
association between genotype and phenotype (2). Spontaneous mutations can occur first in
the proband or be inherited from a parent. There is no gender
predilection and mutations can be splice site, nonsense or deletion
variants. All mutations lead to the insertion of a premature
termination codon (2,17,25).
In the present study, the proband was diagnosed
based on physical examination; and TCS was subsequently confirmed
using molecular analysis. Briefly, one novel heterozygous mutation
in the TCOF1 gene was identified. To the best of our
knowledge, this mutation has not previously been reported in TCS.
The Deafness Variation Database (http://deafnessvariationdatabase.org) and Exome
Aggregation Consortium (http://exac.broadinstitute.org) databases were also
searched and the mutation was not listed. As such, the mutation
described in the present study, chr5:149743675:G>A;
NM_000356.3:c.165-1G>A, appears to be a novel TCOF1
mutation. The mutation is localized in exon 3 of the TCOF1
gene and is a splice site mutation that can lead to exon 3 being
skipped in the matured mRNA. This results in the formation of a
truncated TCOF1 protein with loss of function.
The phenotypic variability observed in TCS makes
diagnosis challenging. Although there is no clear explanation yet,
genotype and phenotype discordance have been suggested by some
studies. In some individuals, phenotypic expression of TCS is so
mild that it is near impossible to diagnose based on physical
examination alone (26). In
contrast, some patients may succumb to respiratory distress soon
after birth due to the severity of TCS symptoms (26). More than 60% of patients with TCS
have no positive family history and the condition is thought to
arise from de novo mutations (5). At present, the reason for the
differences in TCS presentation remains unknown. The wide variation
in symptoms has been attributed to modifier genes, epigenetic
factors and the role of wild-type alleles (26–28).
Some studies have demonstrated that patient phenotype is not
dependent on the type or localization of the mutation responsible
(21,23). Identical mutations in the same family
may cause variable expressivity in different individuals, variable
expressivity of TCOF1 mutations may not be a consequence of
mutational heterogeneity and phenotypic expression of this disorder
may be modified by combined effect of genetic, environmental and
stochastic factors, while there are indications of increased
severity over generations phenotypic expression can not modified by
the gender of the parent (29,30). In
the present study, with the exception of her mother, the proband's
family did not have any features of TCS and no mutations were
identified in the same region. Since the novel TCOF1 gene
mutation was detected in the same region in the mother as in the
proband, it follows that this mutation initially occurred in the
mother and was inherited by the proband with increased
severity.
In conclusion, in the present study a novel
pathogenic mutation of the TCOF1 gene was identified in a
proband with TCS and her mother. The results suggest that this
mutation could be passed on to the next generation in an autosomal
dominant manner. As a result, the progeny of the proband and her
mother have a 50% risk of suffering from TCS and therefore require
genetic counseling.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science foundation of China (grant no. 81030017).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HY and DH contributed to the conception and design
of the study. ZY, YL, YW, XZ, HD, JC performed the experiments. ZY
and YL analyzed the data. ZY prepared the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the General
Hospital of PLA Clinical Human Research Ethics Committee (Beijing,
China). All participants provided written informed consent.
Patient consent for publications
All participants provided written informed
consent.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MD
|
mandibulofacial dysostosis
|
|
TCS
|
Treacher Collins syndrome
|
References
|
1
|
Rosa F, Coutinho MB, Ferreira JP and Sousa
CA: Ear malformations, hearing loss and hearing rehabilitation in
children with Treacher Collins syndrome. Acta Otorrinolaringol Esp.
67:142–147. 2016.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sakai D, Dixon J, Achilleos A, Dixon M and
Trainor PA: Prevention of Treacher Collins syndrome craniofacial
anomalies in mouse models via maternal antioxidant supplementation.
Nat Commun. 7:103282016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luquetti DV, Hing AV, Rieder MJ, Nickerson
DA, Turner EH, Smith J, Park S and Cunningham ML: ‘Mandibulofacial
dysostosis with microcephaly’ caused by EFTUD2 mutations: Expanding
the phenotype. Am J Med Genet A. 161A:1–113. 2013.PubMed/NCBI
|
|
4
|
Vincent M, Collet C, Verloes A, Lambert L,
Herlin C, Blanchet C, Sanchez E, Drunat S, Vigneron J, Laplanche
JL, et al: Large deletions encompassing the TCOF1 and CAMK2A genes
are responsible for Treacher Collins syndrome with intellectual
disability. Eur J Hum Genet. 22:52–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mohan RP, Verma S, Agarwal N and Singh U:
Treacher Collins syndrome: A case report. BMJ Case Rep 2013.
pii:bcr2013009341. 2013.
|
|
6
|
Sakai D and Trainor PA: Treacher Collins
syndrome: Unmasking the role of Tcof1/treacle. Int J Biochem Cell
Biol. 41:1229–1232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mehrotra D, Hasan M, Pandey R and Kumar S:
Clinical spectrum of Treacher Collins syndrome. J Oral Biol
Craniofac Res. 1:36–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Renju R, Varma BR, Kumar SJ and Kumaran P:
Mandibulofacial dysostosis (Treacher Collins syndrome): A case
report and review of literature. Contemp Clin Dent. 5:532–534.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shetty SB, Thomas A and Pidamale R:
Treacher Collins syndrome: A case report and a brief review on
diagnostic aids. Int J Clin Pediatr Dent. 4:235–239. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Andrade EC, Junior VS, Didoni AL, Freitas
PZ, Carneiro AF and Yoshimoto FR: Treacher Collins Syndrome with
choanal atresia: A case report and review of disease features. Braz
J Otorhinolaryngol. 71:107–110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dixon J, Edwards SJ, Anderson I, Brass A,
Scambler PJ and Dixon MJ: Identification of the complete coding
sequence and genomic organization of the Treacher Collins syndrome
gene. Genome Res. 7:223–234. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weiner AM, Scampoli NL and Calcaterra NB:
Fishing the molecular bases of Treacher Collins syndrome. PLoS One.
7:e295742012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Geirdal AØ, Saltnes SS, Storhaug K, Åsten
P, Nordgarden H and Jensen JL: Living with orofacial conditions:
Psychological distress and quality of life in adults affected with
Treacher Collins syndrome, cherubism, or oligodontia/ectodermal
dysplasia-a comparative study. Qual Life Res. 24:927–35. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schaefer E, Collet C, Genevieve D, Vincent
M, Lohmann DR, Sanchez E, Bolender C, Eliot MM, Nürnberg G,
Passos-Bueno MR, et al: Autosomal recessive POLR1D mutation with
decrease of TCOF1 mRNA is responsible for Treacher Collins
syndrome. Genet Med. 16:720–724. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weaver KN, Watt KE, Hufnagel RB, Navajas
Acedo J, Linscott LL, Sund KL, Bender PL, König R, Lourenco CM,
Hehr U, et al: Acrofacial dysostosis, Cincinnati type, a
mandibulofacial dysostosis syndrome with limb anomalies, is caused
by POLR1A dysfunction. Am J Hum Genet. 96:765–774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Noack Watt KE, Achilleos A, Neben CL,
Merrill AE and Trainor PA: The roles of RNA polymerase I and III
subunits Polr1c and Polr1d in craniofacial development and in
Zebrafish models of Treacher Collins syndrome. PLoS Genet.
12:e10061872016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Conte C, D'Apice MR, Rinaldi F,
Gambardella S, Sangiuolo F and Novelli G: Novel mutations of TCOF1
gene in European patients with Treacher Collins syndrome. BMC Med
Genet. 12:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Positional cloning of a gene involved in
the pathogenesis of Treacher Collins syndrome, : The Treacher
Collins syndrome collaborative group. Nat Genet. 12:130–136.
1996.PubMed/NCBI
|
|
19
|
Chang CC and Steinbacher DM: Treacher
Collins syndrome. Semin Plast Surg. 26:83–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dixon J, Jones NC, Sandell LL, Jayasinghe
SM, Crane J, Rey JP, Dixon MJ and Trainor PA: Tcof1/Treacle is
required for neural crest cell formation and proliferation
deficiencies that cause craniofacial abnormalities. Proc Natl Acad
Sci USA. 103:13403–13408. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marszalek-Kruk BA, Wojcicki P, Smigiel R
and Trzeciak WH: Novel insertion in exon 5 of the TCOF1 gene in
twin sisters with Treacher Collins syndrome. J Appl Genet.
53:279–282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
So RB, Gonzales B, Henning D, Dixon J,
Dixon MJ and Valdez BC: Another face of the Treacher Collins
syndrome (TCOF1) gene: Identification of additional exons. Gene.
328:49–57. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Splendore A, Jabs EW, Felix TM and
Passos-Bueno MR: Parental origin of mutations in sporadic cases of
Treacher Collins syndrome. Eur J Hum Genet. 11:718–722. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Splendore A, Silva EO, Alonso LG,
Richieri-Costa A, Alonso N, Rosa A, Carakushanky G, Cavalcanti DP,
Brunoni D and Passos-Bueno MR: High mutation detection rate in
TCOF1 among Treacher Collins syndrome patients reveals clustering
of mutations and 16 novel pathogenic changes. Hum Mutat.
16:315–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trainor PA: Craniofacial birth defects:
The role of neural crest cells in the etiology and pathogenesis of
Treacher Collins syndrome and the potential for prevention. Am J
Med Genet A. 152A:1–2994. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ulusal S, Gurkan H, Vatansever U, Kurkcu
K, Tozkir H and Acunas B: A case of Treacher Collins syndrome.
Balkan J Med Genet. 16:77–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Horiuchi K, Ariga T, Fujioka H, Kawashima
K, Yamamoto Y, Igawa H, Sugihara T and Sakiyama Y: Mutational
analysis of the TCOF1 gene in 11 Japanese patients with Treacher
Collins Syndrome and mechanism of mutagenesis. Am J Med Genet A.
134:363–367. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schlump JU, Stein A, Hehr U, Karen T,
Möller-Hartmann C, Elcioglu NH, Bogdanova N, Woike HF, Lohmann DR,
Felderhoff-Mueser U, et al: Treacher Collins syndrome: Clinical
implications for the paediatrician-a new mutation in a severely
affected newborn and comparison with three further patients with
the same mutation, and review of the literature. Eur J Pediatr.
171:1611–1618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geirdal AO, Saltnes SS, Storhaug K, Asten
P, Nordgarden H and Jensen JL: Living with orofacial conditions:
Psychological distress and quality of life in adults affected with
Treacher Collins syndrome, cherubism, or oligodontia/ectodermal
dysplasia-a comparative study. Qual Life Res. 24:927–935. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Teber OA, Gillessen-Kaesbach G, Fischer S,
Böhringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E,
Hagedorn-Greiwe M, Hammans C, et al: Genotyping in 46 patients with
tentative diagnosis of Treacher Collins syndrome revealed
unexpected phenotypic variation. Eur J Hum Genet. 12:879–890. 2004.
View Article : Google Scholar : PubMed/NCBI
|