Introduction
Guillain-Barre Syndrome (GBS) is a rare
immune-mediated acute inflammatory demyelinating polyneuropathy
with a rapid onset (1). The annual
incidence rate of GBS ranges from 0.81-1.89 per 100,000 people
(2) and males are more frequently
affected than females (3). Previous
studies investigating GBS have focused on assessing objective
parameters, including medical history, physical, laboratory and
electrophysiological examinations (4).
Clinical features of GBS include areflexia and limb
weakness, with additional sensory loss being reported in certain
cases (5). Symptoms can further
develop into neuromuscular paralysis, implicating bulbar, facial
and respiratory function with maximum severity in the initial 2-4
weeks of onset (5). Multiple cranial
neuropathies are a rare variant of GBS, accounting for 5% of
patients (6). Owing to the rarity of
features, this GBS variant is not often considered in the
differential diagnosis due to its lack of progression to limb
weakness (7). To date, there are few
studies that describe pure cranial neuropathy with areflexia and
normal motor and sensory functions as a rare GBA subtype (7,8).
Previous reports also described GBS presented with cranial nerve
damage as the initial symptom (9–11).
Demyelination is often an initial sign of GBS with subsequent
damage to the facial, oculomotor, glossopharyngeal and vagus
nerves. Although the clinical course, severity and prognosis of the
different GBS subtypes are variable, intravenous immunoglobulin
(IVIG) and plasma exchange are proven to be effective treatments
(5). To the best of our knowledge,
the current study describes the first reported case of GBS with
cranial nerve impairment, in which visual impairment was the
initial symptom.
Case report
The present case report was approved by the Ethics
Committee of Baoji Municipal Central Hospital (Baoji, China) and
the patient provided written informed consent.
A 43-year-old female presented with binocular vision
loss for five days and was admitted to the Department of Neurology,
Baoji Municipal Central Hospital on December 20th 2016 for
potential cranial neuropathy. During the initial physical
examination, deterioration of near and distant vision was
identified. The patient recently (2 weeks prior) recovered from an
upper respiratory tract infection and the rest of, her medical
history was otherwise unremarkable. The following neurological
exams were normal: Limb muscular tension (grade 5), response to
sensory stimuli, synesthesia, reflexes of the bilateral biceps,
triceps, knees and Achilles tendons and bilateral Babinski (data
not shown) (12). Those
investigations followed the standard protocols of neurological
investigation. On day 3 post-admission, the patient's symptoms
intensified, with worsening of the blurred binocular vision
accompanied by diplopia, bilateral ptosis, speech difficulty,
excessive coughing during drinking, dysphagia, extremity numbness
and weakness of the limbs (data not shown).
A neurological examination revealed bilateral loss
of visual acuity, with 20/30 for the right eye and 20/25 for the
left (data not shown). The following were also abnormal: Weakened
limb muscular tension (grade 3) and limb reflexes, as well as
weakened sensory feeling upon the direct physical examination (data
not shown). However, the deep sensory reflexes and bilateral
Babinski signs were normal (data not shown). Optical coherence
tomography revealed that the peripapillary retinal nerve fiber
layer was thickened on each side, suggesting bilateral edema of the
optic nerves (data not shown).
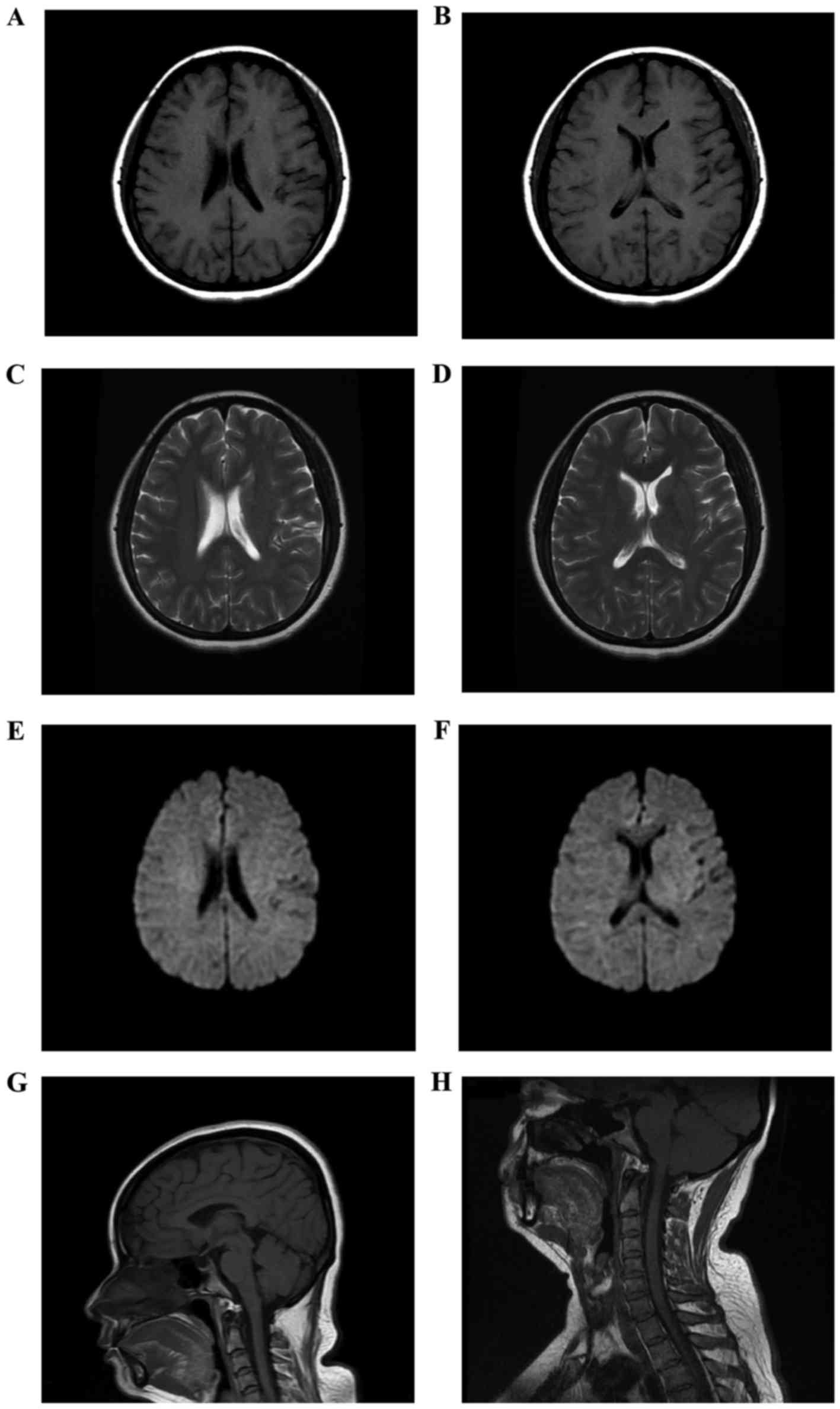
Cerebral magnetic resonance imaging (MRI) and
cervical spinal MRI were normal (Fig.
1). The patient rejected a MRI of the optic nerves due to the
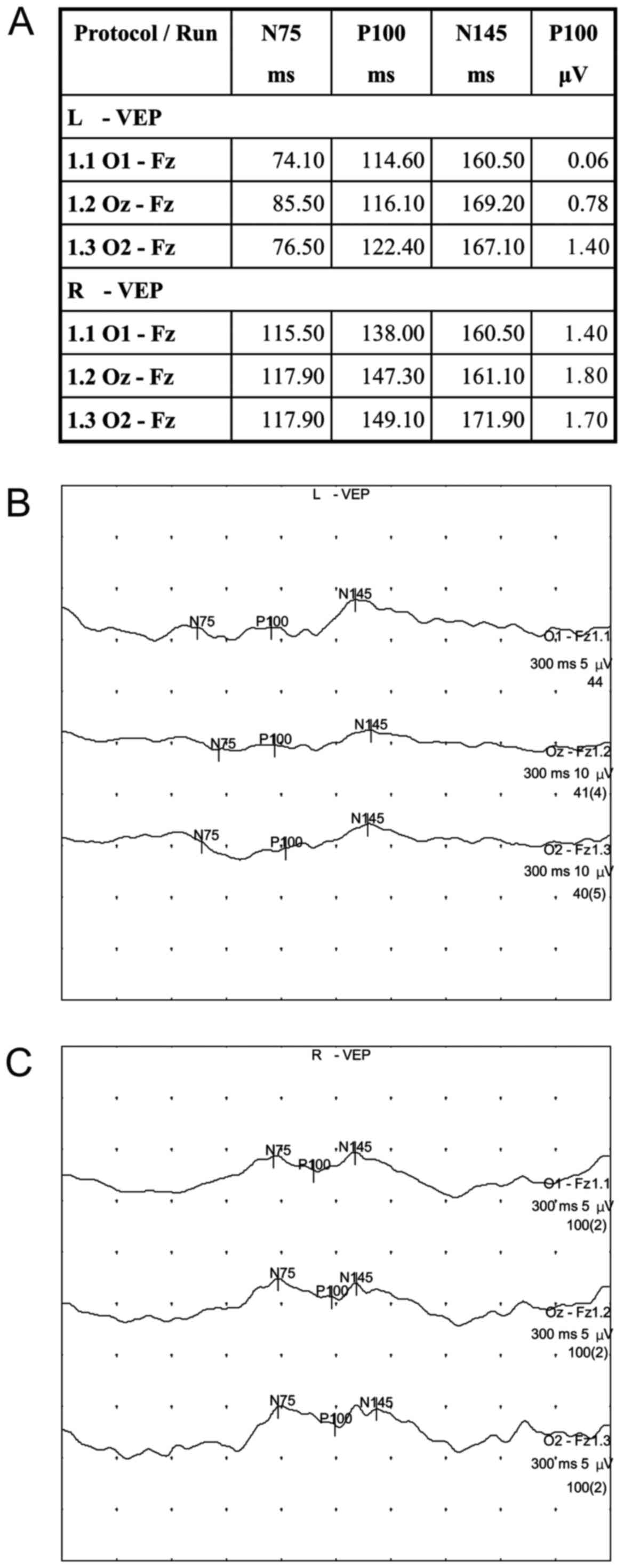
financial burden. Visual evoked potential (VEP) revealed that the
latency of the bilateral full-field P100 was prolonged and the
amplitude was low (Fig. 2).
Electromyography revealed severe impairment of the sensory and
motor fibers in the bilateral median, ulnar, tibial and common
peroneal nerves (Tables I and
II).
 | Table I.Conduction velocity of the sensory
nerves. |
Table I.
Conduction velocity of the sensory
nerves.
|
|
|
|
|
|
| Normal reference
values |
|---|
|
|
|
|
|
|
|
|
|---|
| Limb sensory nerve
conduction | L/R | LAT (m/sec) | AMP (µv) | DN (cm) | NCV (m/sec) | AMP (µv) | NCV (m/sec) |
|---|
| Median | L | 3.17 | 11.20 | 11.60 | 36.90 | 12.60 | 42.10 |
|
| R | 3.18 | 11.30 | 11.00 | 37.60 | 12.50 | 41.70 |
| Ulnar | L | 3.28 | 5.40 | 12.00 | 42.90 | 6.70 | 44.80 |
|
| R | 3.20 | 4.50 | 11.00 | 42.60 | 6.60 | 43.90 |
| Tibial | L | 8.40 | 0.16 | 17.50 | 20.30 | 0.42 | 35.60 |
|
| R | 6.50 | 0.25 | 16.00 | 25.40 | 0.30 | 34.60 |
| Common peroneal | L | 7.19 | 0.24 | 26.00 | 36.90 | 0.60 | 46.39 |
|
| R | 7.80 | 0.21 | 27.00 | 33.80 | 0.60 | 46.53 |
| Table II.Conduction velocity of the motor
nerves. |
Table II.
Conduction velocity of the motor
nerves.
| Limb motor nerve
conduction | L/R | LAT (m/sec) | AMP (mv) | Normal AMP values
(mv) |
|---|
| Median | L | 4.39 | 5.10 | 7.00 |
|
| R | 4.73 | 5.80 | 7.00 |
| Ulnar | L | 3.62 | 5.50 | 7.00 |
|
| R | 3.47 | 5.00 | 7.00 |
| Tibial | L | 6.21 | 2.40 | 4.00 |
|
| R | 6.32 | 2.30 | 4.00 |
| Common peroneal | L | 5.59 | 2.10 | 4.00 |
|
| R | 4.41 | 2.70 | 4.00 |
Next, a lumbar puncture was performed to collect the
cerebrospinal fluid (CSF) 5 days post-admission as previously
described (13). CSF analysis
revealed albuminocytological dissociation and protein-cell
separation (data not shown). The white blood cell count was
0.009×109 cells/l (normal value, 0-0.008×109
cells/l) and the red blood cell count was 0 (normal value, 0-2
individual/HP). Protein, chlorine and sugar concentrations were 862
mg/l (normal value, 0.15-0.45 g/l), 129.6 mmol/l (normal value,
120-130 mmo/l) and sugar 3.69 mmol/l (normal value, 2.5-4.4 mmo/l),
respectively. Analysis of aquaporin-4 immunoglobulin G (AQP4-IgG)
in the peripheral blood and CSF were normal (data not shown). The
peripheral blood anti-Sm antibody, anti-cyclic citrullinated
peptide (anti-CCP) antibodies, rheumatoid factor and neutrophil
cytoplasmic autoantibodies were all normal (data not shown).
Analyses of the clinical symptoms, neurological
examination, electromyography, brain and spinal cord MRI, and
peripheral blood and CSF findings led to a diagnosis of GBS.
Following the identification of GBS, the patient immediately
received IVIG treatment (0.4 g/kg/day). A marked improvement in the
patient's blurred vision, diplopia, ptosis, speech, dysphagia and
limb activities were observed 5 days later. The patient regained
the ability to hold objects in their hands and walk independently.
Additionally, dexamethasone immune modulating therapy (10 mg/day,
once daily) was administrated for 14 days and a neurotrophic
treatment (mecobalamin; 1,000 µg, once daily) was administered for
21 days.
The patient was discharged following 3 weeks when
their blurred vision had returned to normal and with a marked
improvement in her neurological functions. Specifically, the
initially observed diplopia, bilateral ptosis, speech difficulty,
excessive coughing during drinking, dysphagia, extremity numbness
and limb weakness were improved but not completely restored to
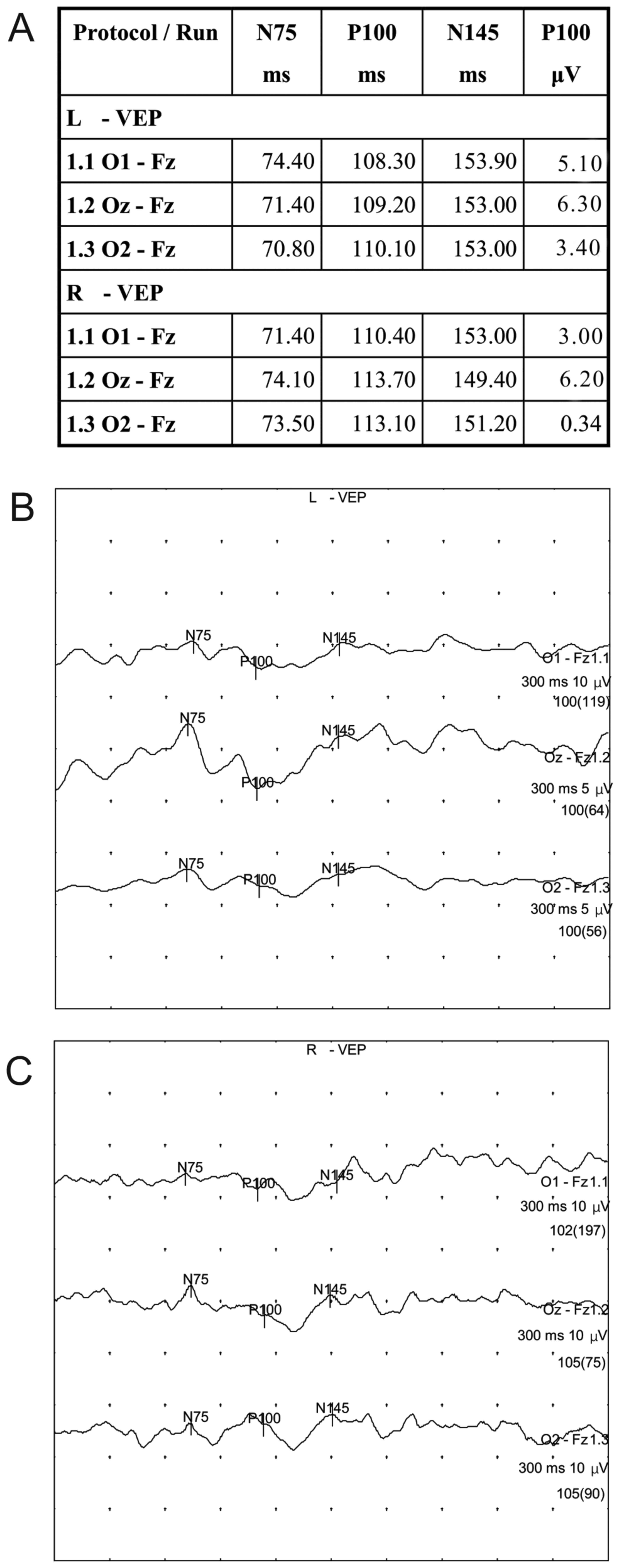
normal. Additionally, the VEP revealed that the latency and the
amplitude of the bilateral full-field P100, N75 and N145 had
returned to normal levels (Fig. 3)
and the patient was able to live independently.
Discussion
GBS is an autoimmune disease caused by peripheral
nerve demyelination. In the present study, a rare case of GBS was
reported in which the patient presented with cranial nerve damage
and visual deterioration as the initial symptom.
In 1966, Morley and Reynolds (14) drew attention to the possibility of
optic neuritis associated with GBS. In 2015, a 10-year-old child
with GBS developed bilateral optic neuritis during the later stages
of the illness (15). Similarly, a
14-year-old male with GBS presented with hyperreflexia and
bilateral papillitis (16). In the
present case, the initial symptoms of GBS were binocular visual
deterioration followed by the development of diplopia and ptosis,
which indicated a lesion involving the bilateral oculomotor nerve.
Subsequently, the patient's inarticulacy and swallowing
difficulties were indicative of a lesion involving the
glossopharyngeal and vagus nerves. Extremity numbness and weakness
of the limbs indicated a lesion involving the peripheral
nerves.
Following 5 days of IVIG therapy, the above clinical
symptoms markedly improved. This was attributed to the ability of
IVIG treatment to adjust humoral immunity and inhibit the
production of autoantibodies, thereby reducing complement-mediated
impairment (17). In addition, the
patient demonstrated improved visual acuity, diplopia and ptosis,
which suggests the therapeutic efficacy of IVIG treatment in GBS
cases involving cranial nerve damage consistent with previously
published data (18).
Atypical cases of GBS may be easily misdiagnosed,
therefore, I recommend that suspected GBS should be confirmed by
the collective analysis of clinical symptoms, neurological
examination, electromyography, brain and spinal cord MRI and CSF
examination. These examinations should provide an accurate
diagnosis that distinguishes GBS from other intracranial diseases,
including brain stem encephalitis, pharyngeal cervical arm
powerlessness and acute ophthalmoplegia (7,19).
Similarly, it is necessary to exclude systemic lupus erythematosus
(20), rheumatoid arthritis
(21), antiphospholipid antibody
syndrome (22) and neuromyelitis
optica (23), which may also
potentially cause nervous system damage.
In the present study, the patient's symptoms
progressed rapidly, which eliminated the possibility of nervous
system damage due to rheumatoid arthritis. In addition, the patient
was previously healthy and had no clinical manifestations,
including body rash, kidney damage, joint pain in the extremities,
pulmonary infection or heart disease. The level of anti-Sm antibody
was normal, which excludes nervous system damage caused by systemic
lupus erythematosus. Additionally, the patient exhibited no
symptoms of headache, vomiting, limb twitching or disturbance of
consciousness and results of a routine blood examination,
anti-cardiolipin antibodies and anti-neutrophil cytoplasmic
antibodies in the serum were normal. The cerebral and cervical MRI
revealed no abnormal signals, which excluded possible nervous
system damage caused by anti-phospholipid antibody syndrome or
granulomatous angiitis. Although the patient presented with vision
loss and signs of optic nerve damage, the absence of sphincter
disturbance and normal findings in the cervical MRI, as well as the
AQP4-IgG and CSF, led to the elimination of neuromyelitis optica
from the differential diagnosis.
In conclusion, the present study reports a rare case
of GBS associated with cranial nerve damage where the initial
presenting symptom was sudden deterioration of binocular vision. I
hypothesize that the patient developed GBS subsequent to a recent
upper respiratory tract infection. The diagnosis of GBS was based
on clinical symptoms, neurological examinations, electromyography,
brain and spinal cord MRI and CSF examination. In addition, the
curative efficacy of IVIG therapy in GBS treatment was
demonstrated. IVIG has multiple roles in the immune system,
including the provision of anti-idiotypic antibodies, blocking
macrophage Fc receptors, inhibiting complement activity and
modulating central B- and T-cell function (24). Due to the autoimmune nature of GBS,
immunoglobulin therapy has been proposed as a potentially effective
treatment (18). Nevertheless, to
the best of my knowledge, the addition of immune-modulatory agents
(dexamethasone) and neurotrophic treatment (mecobalamin) to IVIG
treatment enhanced the therapeutic efficiency.
Acknowledgements
The author would like to thank Dr Jun-Wen Wang,
Department of Neurology, Baoji Municipal Central Hospital, 8
Jiangtan Road, Baoji, Shaanxi 721008, P.R. China for his technical
assistance in performing the present study.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Author contribution
Hui-Jun Wen conceived, designed and drafted the
manuscript, was responsible for the acquisition analysis, revision
and interpretation of data and approved of the final version to be
published. Hui-Jun Wen agrees to be accountable for all aspects of
the work in ensuring that questions related to the accuracy or
integrity of any part of the work.
Ethics approval and consent to
participate
The present case report was approved by the Ethics
Committee of Baoji Municipal Central Hospital (Baoji, China) and
the patient provided written informed consent.
Patient consent for publication
Written informed consent was obtained for
publication.
Competing interests
The author declares that he has no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GBS
|
Guillain-Barre syndrome
|
|
IVIG
|
intravenous immunoglobulin
|
|
MRI
|
magnetic resonance imaging
|
|
VEP
|
visual evoked potential
|
References
|
1
|
Thomas NH: Diagnosis and management of
Guillain-Barré syndrome. Curr Paediatr. 15:287–291. 2005.
View Article : Google Scholar
|
|
2
|
Sejvar JJ, Baughman AL, Wise M and Morgan
OW: Population incidence of Guillain-Barré syndrome: A systematic
review and meta-analysis. Neuroepidemiology. 36:123–133. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bogliun G and Beghi E; Italian GBS
Registry Study Group, : Incidence and clinical features of acute
inflammatory polyradiculoneuropathy in Lombardy, Italy, 1996. Acta
Neurol Scand. 110:100–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fokke C, van den Berg B, Drenthen J,
Walgaard C, van Doorn PA and Jacobs BC: Diagnosis of Guillain-Barré
syndrome and validation of Brighton criteria. Brain. 137:33–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Doorn PA, Ruts L and Jacobs BC:
Clinical features, pathogenesis, and treatment of Guillain-Barré
syndrome. Lancet Neurol. 7:939–950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin JJ, Hsia SH, Wang HS, Lyu RK, Chou ML,
Hung PC, Hsieh MY and Lin KL: Clinical variants of Guillain-Barré
syndrome in children. Pediatr Neurol. 47:91–96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu JY, Jung HY, Kim CH, Kim HS and Kim MO:
Multiple cranial neuropathies without limb involvements:
Guillain-Barre syndrome variant. Ann Rehabil Med. 37:740–744. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lyu RK and Chen ST: Acute multiple cranial
neuropathy. A variant of Guillain-Barré syndrome. Muscle Nerve.
30:433–436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sudulagunta SR, Sodalagunta MB, Sepehrar
M, Khorram H, Bangalore Raja SK, Kothandapani S, Noroozpour Z,
Aheta Sham M, Prasad N, Sunny SP, et al: Guillain-Barré syndrome:
Clinical profile and management. Ger Med Sci. 13:Doc16. 2015.
|
|
10
|
van den Berg B, Walgaard C, Drenthen J,
Fokke C, Jacobs BC and van Doorn PA: Guillain-Barré syndrome:
Pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol.
10:469–482. 2010. View Article : Google Scholar
|
|
11
|
Rafii MS: Case 14: A woman with bilateral
Bell's palsy. Med Gen Med. 8:232006.
|
|
12
|
Bogousslavsky J and Fisher M: Textbook of
neurology. Butterworth-Heinemann Co; pp. 1–18. 1998
|
|
13
|
Peskind ER, Riekse R, Quinn JF, Kaye J,
Clark CM, Farlow MR, Decarli C, Chabal C, Vavrek D, Raskind MA and
Galasko D: Safety and acceptability of the research lumbar
puncture. Alzheimer Dis Assoc Disord. 19:220–225. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morley JB and Reynolds EH: Papilloedema
and the Landry-Guillain-Barré syndrome. Case reports and a review.
Brain. 89:205–222. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deshmukh IS, Bang AB, Jain MA and Vilhekar
KY: Concurrent acute disseminated encephalomyelitis and
Guillain-Barre syndrome in a child. J Pediatr. Neurosci. 10:61–63.
2015.
|
|
16
|
Incecik F, Herguner OM, Besen S, Yar K and
Altunbasak S: Guillain-Barré syndrome with hyperreflexia and
bilateral papillitis in a child. J Pediatr Neurosci. 11:71–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hughes RA, Swan AV and van Doorn PA:
Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane
Database Syst Rev: CD002063. 2014. View Article : Google Scholar
|
|
18
|
Raphaël JC, Chevret S, Hughes RA and
Annane D: Plasma exchange for Guillain-Barré syndrome. Cochrane
Database Syst Rev: CD001798. 2012. View Article : Google Scholar
|
|
19
|
Edvardsson B and Persson S: Polyneuritis
cranialis presenting with anti-GQ1b IgG antibody. J Neurol Sci.
281:125–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ali N, Rampure R, Malik F, Jafri SI and
Amberker D: Guillain-Barré syndrome occurring synchronously with
systemic lupus erythematosus as initial manifestation treated
successfully with low-dose cyclophosphamide. J Community Hosp
Intern Med Perspect. 6:306892016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silburn S, McIvor E, McEntegart A and
Wilson H: Guillain-Barré syndrome in a patient receiving
anti-tumour necrosis factor alpha for rheumatoid arthritis: A case
report and discussion of literature. Ann Rheum Dis. 67:575–576.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hara M: Intravenous immunoglobulin (IVIG).
Nihon Rinsho. 67:599–605. 2009.(In Japanese). PubMed/NCBI
|
|
23
|
Kusunoki S: Neuroimmunological diseases:
Update. Nihon Rinsho. 71:771–777. 2013.(In Japanese). PubMed/NCBI
|
|
24
|
Hughes RA: Intravenous immunoglobulin for
chronic inflammatory demyelinating polyradiculoneuropathy: The ICE
trial. Expert Rev Neurother. 9:789–795. 2009. View Article : Google Scholar : PubMed/NCBI
|