Introduction
Congenital cataract is an important cause of
blindness in children globally (1).
A total of 10.7–14.0% of the affected children are blind (1). This lens disease exhibits clinical and
genetic heterogeneity; autosomal dominant inheritance is the most
common. Currently, an increasing number of genes have been
identified as associated with various forms of congenital
cataracts. These genes include crystallin genes [crystallin α A
(CRYAA) (2), crystallin α B (CRYAB)
(3), crystallin A1/A3 (CRYBA1/A3)
(4), crystallin A4 (CRYBA4)
(5), crystallin B1 (CRYBB1)
(6), crystallin B2 (CRYBB2)
(7), crystallin B2 (CRYBB3)
(8), crystallin γ C (CRYGC)
(9), crystallin γ D (CRYGD)
(10) and crystallin γ S (CRYGS)
(11)], transcription factors [heat
shock transcription factor 4 (12),
paired-like homeodomain transcription factor 3 (13) and MAF bZIP transcription factor
(14)], skeleton protein genes
[beaded filament structural proteins 1 (15) and 2 (16)], membrane transporter genes [major
intrinsic protein of lens fiber (MIP) (17), gap junction protein α 8 (GJA8)
(18), gap junction protein α 3
(GJA3) (19) and lens intrinsic
membrane protein 2 (20)],
glucosaminyl (N-acetyl) transferase 2 (21), charged multivesicular body protein 4B
(22), and transmembrane protein 114
(23). Elucidating the structure and
functional characteristics of these candidate genes and their
protein products may aid in understanding the occurrence of
cataracts, and the functional and structural implications of their
mutations may provide important clues for understanding the disease
etiology. In the present study, a heterozygous c.97C>T
transition mutation of the MIP gene was identified in a family from
the Chinese Guangxi Zhuang Autonomous Region with congenital
nuclear cataract. The mutation completely cosegregated with the
disease. This is the first cataract-associated mutation identified
among patients of Guangxi Zhuang ethnicity.
Materials and methods
Clinical data and sample
collection
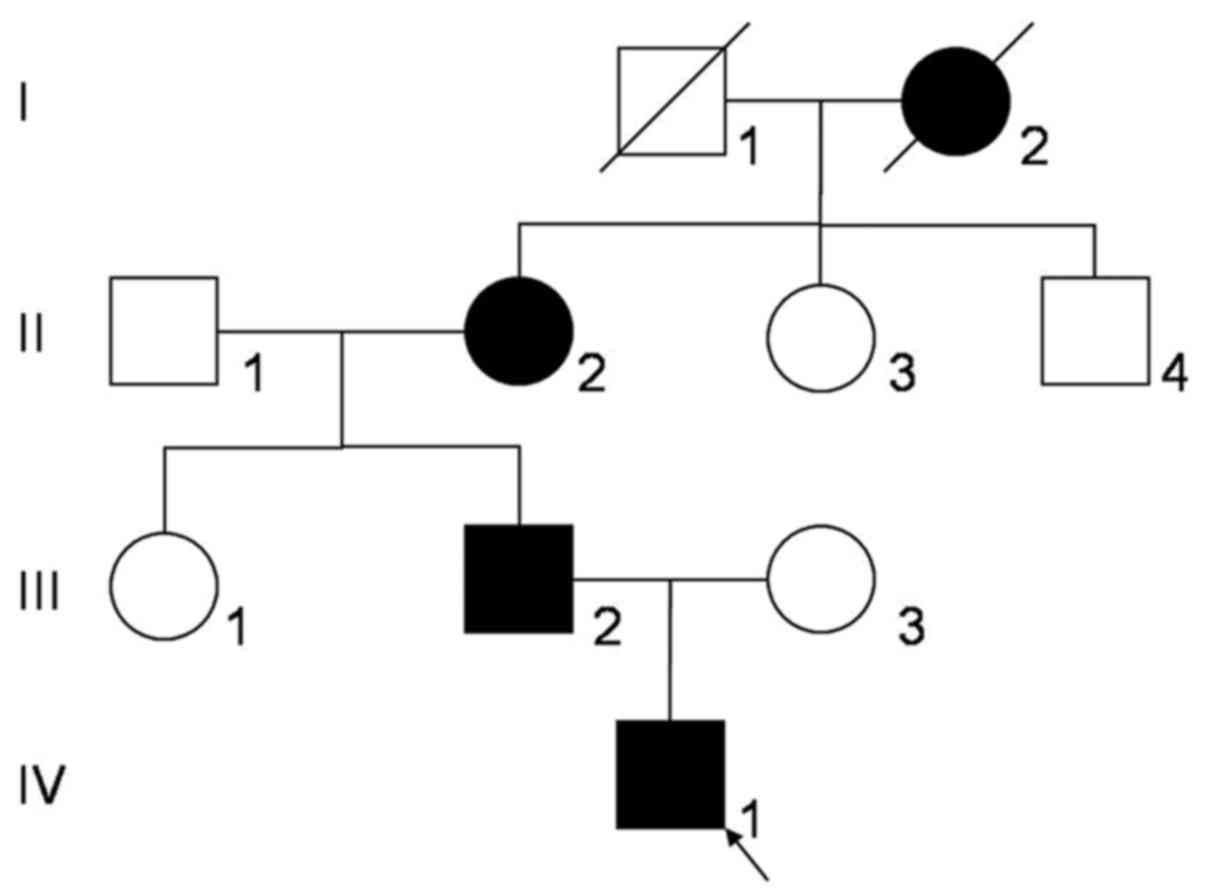
A three-generation Chinese Zhuang family (Fig. 1) with congenital nuclear cataract was
recruited from the People's Hospital of Guangxi Zhuang Autonomous
Region (Nanning, China). The study included eight family members,
including three affected individuals (II:2, III:2 and IV:1) and
five unaffected individuals (II:1, II:3, II:4, III:1 and III:3).
All participants underwent physical and ophthalmic examinations. An
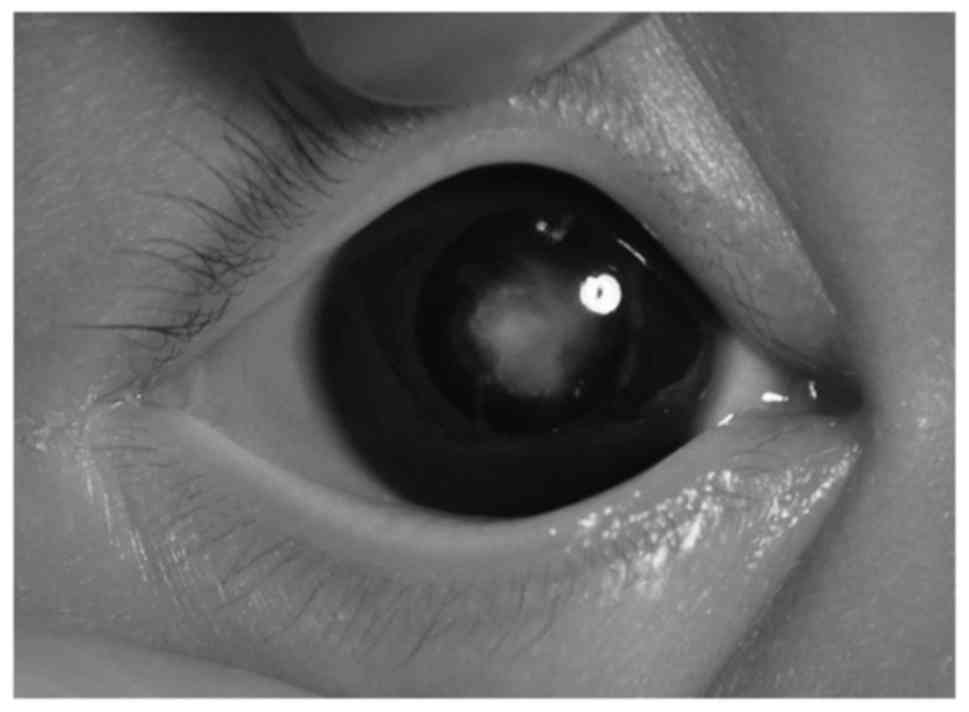
image of the lens opacity of the proband was captured (Fig. 2). A total of 100 Guangxi Zhuang
ethnicity subjects without congenital cataract were recruited as
normal controls. All patients included in the present study
provided written informed consent for participation and
publication. A total of 5 ml of venous blood was collected from
family members and controls using BD Vacutainer® Blood
Collection tubes (BD Biosciences, San Jose, CA, USA) containing
EDTA. Genomic DNA was extracted by QIAamp DNA Blood kits (Qiagen
Sciences, Inc., Gaithersburg, MD, USA). The present study was
approved by the Institutional Review Committee of the People's
Hospital of Guangxi Zhuang Autonomous Region and followed the
provisions of the Declaration of Helsinki.
Mutation detection
Known protein coding regions of candidate genes
associated with autosomal dominant congenital cataract, including
CRYAA, CRYAB, CRYBA1, CRYBB2, CRYGC, CRYGD, CRYGS, GJA3, GJA8 and
MIP, were amplified using polymerase chain reaction (PCR). The
primer sequences were listed in Table
I. The PCR mixtures was as follows: 12.5 µl 2 ×Taq PCR
Mastermix (Tiangen Biotech Co., Ltd., Beijing, China), 1 µl forward
primer, 1 µl reverse primer, 1 µl genomic DNA and ddH2O
up to a volume of 25 µl. PCR cycling conditions consisted of the
following: An initial denaturation at 94°C for 7 min, 40 cycles of
denaturation at 94°C for 30 sec, annealing at 62°C for 30 sec and
extension at 72°C for 45 sec, then a final extension at 72°C for 8
min and a last hold at 4°C.
 | Table I.The primers used for PCR. |
Table I.
The primers used for PCR.
|
| Primer sequence
(5′→3′) |
|
|---|
|
|
|
|
|---|
| Name | Forward | Reverse | Product length (base
pair) |
|---|
| CRYAA-1 |
AGCAGCCTTCTTCATGAGC |
CAAGACCAGAGTCCATCG | 584 |
| CRYAA-2 |
GGCAGGTGACCGAAGCATC |
GAAGGCATGGTGCAGGTG | 550 |
| CRYAA-3 |
GCAGCTTCTCTGGCATGG |
GGGAAGCAAAGGAAGACAGA | 511 |
| CRYAB-1 |
AACCCCTGACATCACCATTC |
AAGGACTCTCCCGTCCTAGC | 250 |
| CRYAB-2 |
CCATCCCATTCCCTTACCTT |
GCCTCCAAAGCTGATAGCAC | 350 |
| CRYAB-3 |
TCTCTCTGCCTCTTTCCTCA |
CCTTGGAGCCCTCTAAATCA | 400 |
| CRYBA1-1 |
GGCAGAGGGAGAGCAGAGTG |
CACTAGGCAGGAGAACTGGG | 550 |
| CRYBA1-2 |
AGTGAGCAGCAGAGCCAGAA |
GGTCAGTCACTGCCTTATGG | 508 |
| CRYBA1-3 |
AAGCACAGAGTCAGACTGAAGT |
CCCCTGTCTGAAGGGACCTG | 463 |
| CRYBA1-4 |
GTACAGCTCTACTGGGATTG |
ACTGATGATAAATAGCATGAACG | 355 |
| CRYBA1-5 |
GAATGATAGCCATAGCACTAG |
TACCGATACGTATGAAATCTGA | 597 |
| CRYBA1-6 |
CATCTCATACCATTGTGTTGAG |
CATCTCATACCATTGTGTTGAG | 528 |
| CRYBB2-1 |
GTTTGGGGCCAGAGGGGAGTGGT |
TGGGCTGGGGAGGGACTTTCAGTA | 350 |
| CRYBB2-2 |
CCTTCAGCATCCTTTGGGTTCTCT |
GCAGTTCTAAAAGCTTCATCAGTC | 330 |
| CRYBB2-3 |
GTAGCCAGGATTCTGCCATAGGAA |
GTGCCCTCTGGAGCATTTCATAGT | 360 |
| CRYBB2-4 |
GGCCCCCTCACCCATACTCA |
CTTCCCTCCTGCCTCAACCTAATC | 230 |
| CRYBB2-5 |
CTTACCCTTGGGAAGTGGCAATGG |
TCAAAGACCCACAGCAGACAAGTT | 600 |
| CRYGC-1 |
TGCATAAAATCCCCTTACCG |
CCTCCCTGTAACCCACATTG | 514 |
| CRYGC-2 |
TGGTTGGACAAATTCTGGAAG |
CCCACCCCATTCACTTCTTA | 430 |
| CRYGD-1 |
CAGCAGCCCTCCTGCTAT |
GGGTCCTGACTTGAGGATGT | 550 |
| CRYGD-2 |
GCTTTTCTTCTCTTTTTATTTCTGG |
AAGAAAGACACAAGCAAATCAGT | 308 |
| CRYGS-2 |
GAAACCATCAATAGCGTCTAAATG |
TGAAAAGCGGGTAGGCTAAA | 575 |
| CRYGS-3 |
AATTAAGCCACCCAGCTCCT |
GGGAGTACACAGTCCCCAGA | 479 |
| CRYGS-4 |
GACCTGCTGGTGATTTCCAT |
CACTGTGGCGAGCACTGTAT | 974 |
| GJA3-1 |
CGGTGTTCATGAGCATTTTC |
CTCTTCAGCTGCTCCTCCTC | 450 |
| GJA3-2 |
GAGGAGGAGCAGCTGAAGAG |
AGCGGTGTGCGCATAGTAG | 450 |
| GJA3-3 |
TCGGGTTCCCACCCTACTAT |
TATCTGCTGGTGGGAAGTGC | 300 |
| GJA8-1 |
CCGCGTTAGCAAAAACAGAT |
CCTCCATGCGGACGTAGT | 420 |
| GJA8-2 |
GCAGATCATCTTCGTCTCCA |
GGCCACAGACAACATGAACA | 330 |
| GJA8-3 |
CCACGGAGAAAACCATCTTC |
GAGCGTAGGAAGGCAGTGTC | 350 |
| GJA8-4 |
TCGAGGAGAAGATCAGCACA |
GGCTGCTGGCTTTGCTTAG | 500 |
| MIP-1 |
TCTCGGCTCATCTCCCAGTT |
GGCAATAGAGAGACAGGACAC | 635 |
| MIP-2 |
TGAAGGAGCACTGTTAGGAGATG |
AGAGGGATAGGGCAGAGTTGATT | 500 |
| MIP-3 |
CCAGACAGGGCATCAGT |
TGGTACAGCAGCCAACAC | 677 |
| MIP-4 |
AAGGTGTGGGATAAAGGAGT |
TTCTTCATCTAGGGGCTGGC | 389 |
Bioinformatics analysis
PCR products were sequenced by ABI 3730 automated
sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) from both directions. The results of sequencing
were analyzed with Chromas (2.3 edition; Technelysium Pty Ltd,
South Brisbane, Australia) and compared with reference sequences
from the NCBI database (https://www.ncbi.nlm.nih.gov/). Bioinformatics
analysis of wild-type and mutant MIP protein sequences was
conducted using a polymorphism phenotyping v2 (PolyPhen-2) software
(version 2.0; http://genetics.bwh.harvard.edu/pph2/), and the
effects of mutations on biochemical properties were predicted.
PolyPhen-2 was based on position-specific independent counting from
multiple sequence alignments (24),
was used to predict whether the amino acid substitutions affected
the protein function. The hydrophilicity of wild-type and mutant
protein products was analyzed using online biological software
program Misc Protein Analysis (https://fasta.bioch.virginia.edu/fasta_www2/fasta_www.cgi?rm=misc1).
Results
Clinical data
There were 4 affected individuals among the 10
family members (Fig. 1). The proband
(IV:1) was a one-year old male whose great-grandmother (I:2),
grandmother (II:2) and father (III:2) had poor eyesight in their
childhood. Among them, one (I:2) succumbed to mortality and two
(II:2 and III:2) were examined prior to cataract removal. The
proband exhibited a bilateral cataract characterized as a central
nuclear opacity involving the embryonic and fetal nuclei with
posterior polar opacities (Fig. 2).
There was no family history of other eye conditions or systemic
diseases.
Mutation analysis
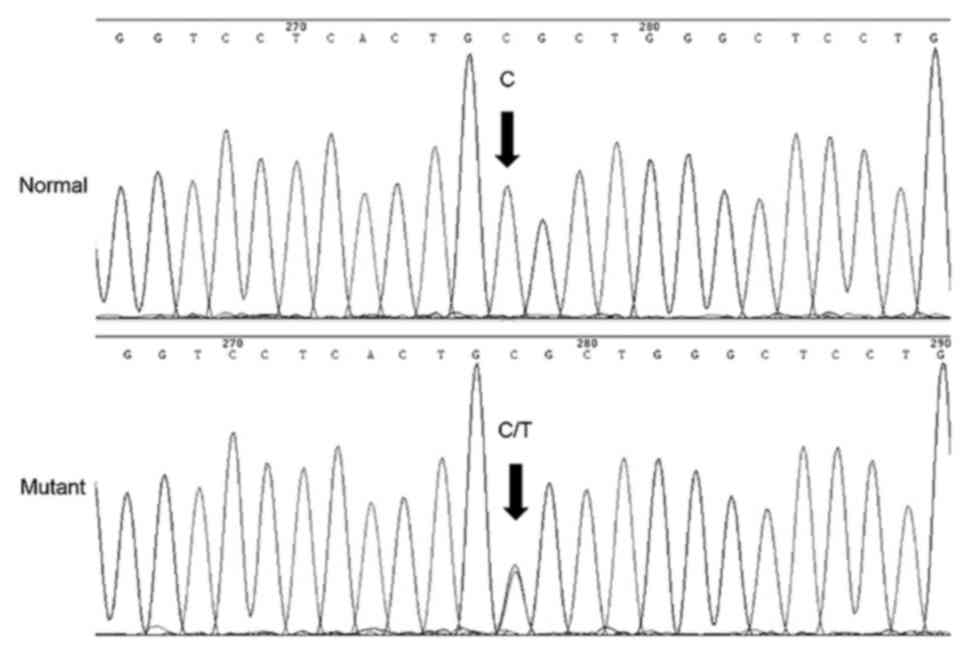
Direct sequencing of the candidate genes indicated
that in the MIP gene position 97, as a result of the C-T
transition, the highly conserved arginine was substituted by
cysteine in the codon 33 (Fig. 3).
This mutation was detected in all affected members, however, it was
not observed in the unaffected family members or normal `controls.
No significant nucleotide polymorphisms were identified in other
candidate genes.
Bioinformatics analysis
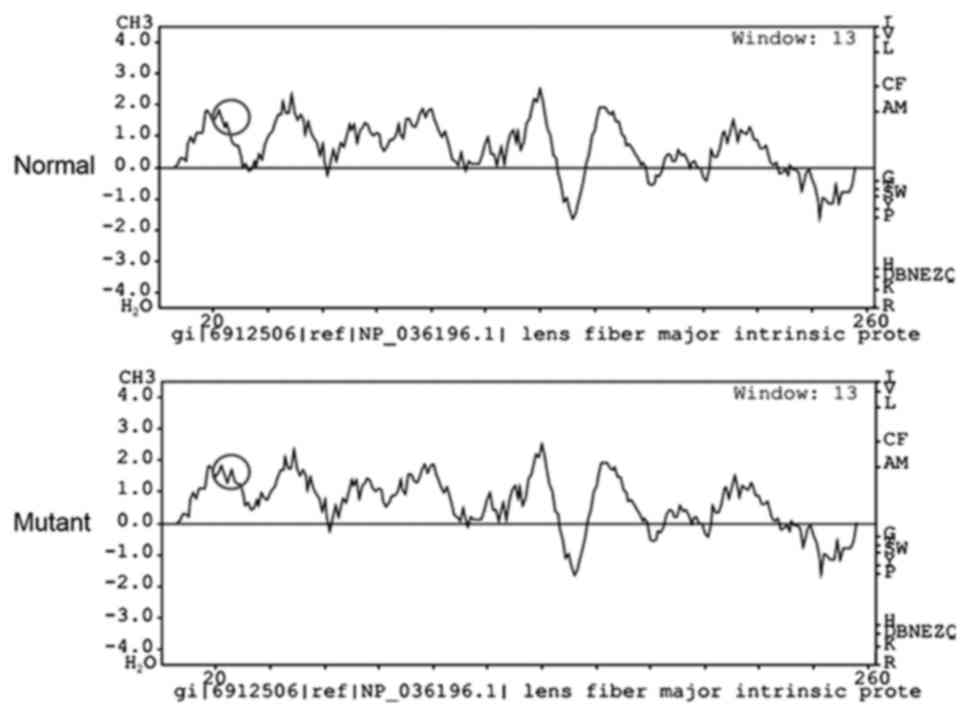
Bioinformatics analysis with PolyPhen-2 revealed
that the replacement in the MIP gene at position 33 from R to C
scored 0.999 (sensitivity, 0.14; specificity, 0.99) and was
predicted as possibly damaging. The hydrophobicity of this variant
was markedly elevated (Fig. 4).
Discussion
MIP is a member of the water-channel family of
proteins. It is the most abundant type of membrane protein in the
mature lens, expressed only in end-stage differentiated fibrous
cells (25). The function of MIP, as
a water channel and an adhesion molecule in the lens fiber,
contributes to the formation of the small intercellular space of
the lens fiber, which is necessary for lens transparency and
adaptation (26).
MIP gene is located on chromosome 12q13 and several
mutations in MIP are associated with human genetic cataracts
(27). At present, 20 different
mutations have been identified to cause human congenital cataracts,
including p.M1T, p.R33C, p.V107I, p.R113X, p.E134G, p.T138R,
p.D150H, p.G165D, p.A169PfsX15, p.L170PfsX31, p.Y177C, p.R187C,
p.N200GfsX12, p.W202X, c.606+1G>A splicing, p.V203fs,
p.G213VfsX46, p.G215D, p.Y219X and p.R233K, and 3 variations have
been found to be associated with age-related cataracts (rs2269348,
rs117788190 and rs74641138) (27).
According to the amino acid sequences, the members of the
water-channel family are predicted to share a common protein
topology consisting of six transmembrane domains and five
extracellular loops (28). Out of
them, the first extracellular loop contains the following residues:
33R, 34W, 35A, 36P, 37G, 38P, 39L and 40H (28). The mutation investigated in the
current study was in the first residue of the first extracellular
loop of the MIP protein.
In the present study, a missense mutation c.97C>T
in MIP gene leading to substitution of arginine with cysteine
(p.R33C) was found in a Chinese family with congenital central
nucleus and posterior polar cataract. This mutation co-segregated
with the disease phenotype and was not found in the 100 unrelated
control individuals. The p.R33C substitution was reported in a
Chinese family (29) and an
Australian sporadic case (30). In
2007, Gu et al (29), was the
first to report the c.97C>T mutation in a Chinese family with an
autosomal dominant total cataract. This was the first reported case
of cataracts caused by a mutation located outside the transmembrane
portion of MIP. The authors hypothesized that the p.R33C
substitution may allow for the formation of intermolecular
disulfide bonds and thereby destabilize the wild-type structure of
MIP. The abnormal formation of the disulfide bonds may affect the
position of MIP in the plasma membranes (29). Ma et al (30) identified p.R33C in an Australia
sporadic congenital cataract case by using the next generation
sequencing technique, which further confirmed the R33C mutation in
MIP is associated with congenital cataract.
In the Chinese and Australian families reported in
the previous studies, the phenotype was described as a full
cataract (29,30). In the present study, the phenotype
was described as a bilateral central nuclear opacity involving
embryonic and fetal lens nuclei and posterior polar opacity. The
family analyzed in the present study was of Guangxi Zhuang
ethnicity, which is the most populous minority in the Guangxi
Zhuang Autonomous Region (31). The
present study provided additional information for the understanding
of the genetic diversity of the Chinese nation. It was demonstrated
that the phenotypic heterogeneity of the p.R33C mutation in MIP may
be associated with the occurrence of congenital cataracts.
The molecular consequence of the p.R33C mutation in
MIP should be further examined to provide an in-depth understanding
of the pathogenesis of congenital cataracts. Additional studies may
examine MIP mutations, which cause cataracts, to gain a greater
understanding of the basis of MIP-mediated cataractogenesis.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Self-Financing Project of Guangxi Zhuang Region Health Department
(grant no. Z2016594).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ analyzed and interpreted the data, and was a
major contributor in writing the manuscript. LLi made substantial
contributions to conception of the study. LLu performed the Genomic
DNA extraction, gel electrophoresis, polymerase chain reaction
analysis, polymerase chain reaction product sequencing and drafted
the manuscript. LM made substantial contributions to design of the
study and acquisition of data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Committee of the People's Hospital of Guangxi Zhuang
Autonomous Region and followed the provisions of the Declaration of
Helsinki. Written informed consent to participate was obtained from
all patients included in the present study.
Patient consent for publication
Written informed consent for publication was
obtained from all patients included in the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gralek M, Kanigowska K and Seroczynska M:
Cataract in children-not only an ophthalmological problem. Med
Wieku Rozwoj. 11:227–230. 2007.(In Polish). PubMed/NCBI
|
|
2
|
Kong XD, Liu N, Shi HR, Dong JM, Zhao ZH,
Liu J, Li-Ling J and Yang YX: A novel 3-base pair deletion of the
CRYAA gene identified in a large Chinese pedigree featuring
autosomal dominant congenital perinuclear cataract. Genet Mol Res.
14:426–432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiao X, Khan SY, Irum B, Khan AO, Wang Q,
Kabir F, Khan AA, Husnain T, Akram J, Riazuddin S, et al: Missense
mutations in CRYAB are liable for recessive congenital cataracts.
PLoS One. 10:e01379732015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khan AO, Aldahmesh MA and Alkuraya FS:
Phenotypes of recessive pediatric cataract in a cohort of children
with identified homozygous gene mutations (an american
ophthalmological society thesis). Trans Am Ophthalmol Soc.
113:T72015.PubMed/NCBI
|
|
5
|
Zhou G, Zhou N, Hu S, Zhao L, Zhang C and
Qi Y: A missense mutation in CRYBA4 associated with congenital
cataract and microcornea. Mol Vis. 16:1019–1024. 2010.PubMed/NCBI
|
|
6
|
Wu Q, Shi H, Liu N, Lu N, Jiang M, Zhao Z
and Kong X: Mutation analysis of CRYBB1 gene and prenatal diagnosis
for a chinese kindred featuring autosomal dominant congenital
nuclear cataract. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 30:266–269.
2013.(In Chinese). PubMed/NCBI
|
|
7
|
Faletra F, d'Adamo AP, Pensiero S,
Athanasakis E, Catalano D, Bruno I and Gasparini P: A novel CRYBB2
missense mutation causing congenital autosomal dominant cataract in
an Italian family. Ophthalmic Genet. 34:115–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li D, Wang S, Ye H, Tang Y, Qiu X, Fan Q,
Rong X, Liu X, Chen Y, Yang J and Lu Y: Distribution of gene
mutations in sporadic congenital cataract in a han chinese
population. Mol Vis. 22:589–598. 2016.PubMed/NCBI
|
|
9
|
Prokudin I, Simons C, Grigg JR, Storen R,
Kumar V, Phua ZY, Smith J, Flaherty M, Davila S and Jamieson RV:
Exome sequencing in developmental eye disease leads to
identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1.
Eur J Hum Genet. 22:907–915. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang G, Chen Z, Zhang W, Liu Z and Zhao J:
Novel mutations in CRYGD are associated with congenital cataracts
in Chinese families. Sci Rep. 6:189122016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang Z, Li Q, Zhu S and Ma X: A G57W
Mutation of CRYGS associated with autosomal dominant pulverulent
cataracts in a chinese family. Ophthalmic Genet. 36:281–283. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Zhang Q, Zhou LX and Tang ZH: A
novel HSF4 mutation in a Chinese family with autosomal dominant
congenital cataract. J Huazhong Univ Sci Technolog Med Sci.
35:316–318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye X, Zhang G, Dong N and Meng Y: Human
pituitary homeobox-3 gene in congenital cataract in a chinese
family. Int J Clin Exp Med. 8:22435–22439. 2015.PubMed/NCBI
|
|
14
|
Narumi Y, Nishina S, Tokimitsu M, Aoki Y,
Kosaki R, Wakui K, Azuma N, Murata T, Takada F, Fukushima Y and
Kosho T: Identification of a novel missense mutation of MAF in a
Japanese family with congenital cataract by whole exome sequencing:
A clinical report and review of literature. Am J Med Genet A.
164A:1272–1276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Zhang T, Wu D and Zhang J: A novel
beaded filament structural protein 1 (BFSP1) gene mutation
associated with autosomal dominant congenital cataract in a chinese
family. Mol Vis. 19:2590–2595. 2013.PubMed/NCBI
|
|
16
|
Liu Q, Wang KJ and Zhu SQ: A novel p.G112E
mutation in BFSP2 associated with autosomal dominant pulverulent
cataract with sutural opacities. Curr Eye Res. 39:1013–1019. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin L, Guo L, Wang H, Li T, Lou G, Guo Q,
Hou Q, Liu H, Liao S and Liu Z: A novel MIP mutation in familial
congenital nuclear cataracts. Eur J Med Genet. 59:488–491. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu Y, Yu H, Wang W, Gong X and Yao K: A
novel GJA8 mutation (p.V44A) causing autosomal dominant congenital
cataract. PLoS One. 9:e1154062014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li B, Liu Y, Liu Y, Guo H, Hu Z, Xia K and
Jin X: Identification of a GJA3 mutation in a large family with
bilateral congenital cataract. DNA Cell Biol. 35:135–139. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pras E, Levy-Nissenbaum E, Bakhan T, Lahat
H, Assia E, Geffen-Carmi N, Frydman M, Goldman B and Pras E: A
missense mutation in the LIM2 gene is associated with autosomal
recessive presenile cataract in an inbred Iraqi Jewish family. Am J
Hum Genet. 70:1363–1367. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Happ H, Weh E, Costakos D, Reis LM and
Semina EV: Case report of homozygous deletion involving the first
coding exons of GCNT2 isoforms A and B and part of the upstream
region of TFAP2A in congenital cataract. BMC Med Genet. 17:642016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shiels A, Bennett TM, Knopf HL, Yamada K,
Yoshiura K, Niikawa N, Shim S and Hanson PI: CHMP4B a novel gene
for autosomal dominant cataracts linked to chromosome 20q. Am J Hum
Genet. 81:596–606. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jamieson RV, Farrar N, Stewart K, Perveen
R, Mihelec M, Carette M, Grigg JR, McAvoy JW, Lovicu FJ, Tam PP, et
al: Characterization of a familial t(16;22) balanced translocation
associated with congenital cataract leads to identification of a
novel gene TMEM114 expressed in the lens and disrupted by the
translocation. Hum Mutat. 28:968–977. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gonen T, Cheng Y, Kistler J and Walz T:
Aquaporin-0 membrane junctions form upon proteolytic cleavage. J
Mol Biol. 342:1337–1345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chepelinsky AB: Structural function of
MIP/aquaporin 0 in the eye lens; genetic defects lead to congenital
inherited cataracts. Handb Exp Pharmacol. 265–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shiels A, Bennett TM and Hejtmancik JF:
Cat-Map: Putting cataract on the map. Mol Vis. 16:2007–2015.
2010.PubMed/NCBI
|
|
28
|
Varadaraj K, Kumari SS, Patil R, Wax MB
and Mathias RT: Functional characterization of a human aquaporin 0
mutation that leads to a congenital dominant lens cataract. Exp Eye
Res. 87:9–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu F, Zhai H, Li D, Zhao L, Li C, Huang S
and Ma X: A novel mutation in major intrinsic protein of the lens
gene (MIP) underlies autosomal dominant cataract in a Chinese
family. Mol Vis. 13:1651–1656. 2007.PubMed/NCBI
|
|
30
|
Ma AS, Grigg JR, Ho G, Prokudin I,
Farnsworth E, Holman K, Cheng A, Billson FA, Martin F, Fraser C, et
al: Sporadic and familial congenital cataracts: Mutational spectrum
and new diagnoses using next-generation sequencing. Hum Mutat.
37:371–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guangxi Statistics Bureau: Main data
bulletin of the sixth national census in Guangxi, . 2010.Guangxi
Statistics Bureau. http://www.gxtj.gov.cn/tjsj/tjgb/rkpc/201107/t20110701_2168.htmlNovember
1st, 2010.
|